Синдром Шерешевского-Тернера

Синдром Шерешевского-Тернера представляет собой генетическое заболевание, обусловленное отсутствием половой Х-хромосомы или структурными изменениями в ней. Патология в подавляющем большинстве случаев выявляется у девочек, у мальчиков — в крайне редких случаях. Синдром диагностируется примерно у одного из 2500-4500 новорожденных.
Суть патологии заключается в том, что имеет место полная утрата Х-хромосомы или же структурные изменения в ней при нормальном кариотипе, что провоцирует аномалии развития половых желез на ранних этапах развития эмбриона.
Причины развития синдрома
Хромосомные аномалии могут быть обусловлены как генетическим фактором, так и воздействием внешних факторов, таких как ионизирующее излучение или токсические вещества.
Симптомы
В ряде случаев какие-либо характерные симптомы, позволяющие заподозрить синдром Шерешевского-Тернера у новорожденного, могут почти полностью отсутствовать. Однако нередко обнаруживаются внешние специфические признаки, к которым можно отнести:
- Крыловидные складки кожи на шее
- Соски широко расставлены и втянуты
- Отечные конечности (стопы и кисти)
- Низкие показатели роста и веса ребенка при доношенной беременности
В дальнейшем у пациентов с синдромом Шерешевского-Тернера отмечается задержка в росте, что становится все более очевидным уже к подростковому возрасту и составляет у взрослого порядка 25-ти сантиметров.
Среди внешних признаков стоит отметить так называемое «лицо сфинкса» (миопатическое лицо) с вялой мимикой и отсутствием складок на лбу, немного приоткрытым ртом и глазами, которые не полностью закрываются.
В подростковом возрасте у девочек отмечается задержка полового развития, которая характеризуется первичной аменореей, недоразвитием вторичных половых признаков (в частности молочных желез, роста волос на в подмышечных впадинах и на лобке).
Нередко у пациентов с синдромом Шерешевского-Тернера выявляются пороки развития различных органов и систем организма: патологии опорно-двигательного аппарата, сердечно-сосудистой, мочевыделительной системы, органов слуха.
Возможно отставание в интеллектуальном развитии, эмоциональная нестабильность, повышенная тревожность, депрессивные состояния.
Наличие и степень проявления перечисленных симптомов зависят от вариантов заболевания, связанных с характером генетической патологии.
Хотите записаться на прием?
Кариотип при синдроме Шерешевского-Тернера
У пациенток могут определяться различные кариотипы.
Кариотип 45Х
Примерно у половины женщин, страдающих этим синдромом, в результате исследования определяется кариотип 45Х, что свидетельствует об утрате одной хромосомы (в 75% случаев обнаруживается отсутствие хромосомы, наследуемой с отцовской стороны).
Влагалище и матка не обнаруживаются, в то же время в малом тазу присутствуют тяжи, аналогичные тем, которые обнаруживаются при синдроме Рокитанского-Кюстнера. У пациенток с таким кариотипом на месте яичников определяются соединительнотканные тяжи. Ткань яичников полностью отсутствует.
Обследование выявляет повышенный уровень гонадотропных гормонов, в частности ФСГ (фолликулостимулирующего гормона). Выражены внешние характерные признаки, часто имеют место нарушения со стороны различных систем организма.
Мозаичный кариотип
Помимо этого, может быть определен мозаичный кариотип (наличие клеток с различным кариотипом), такой например, как 45Х/46XY или 45Х/46ХХ. В зависимости от мозаичного варианта и количества измененных клеток варьируются проявления синдрома.
Если у женщины определяется кариотип 45Х/46ХХ, то присутствуют яичники, размер которых уменьшен, и матка без аномалий развития.
Для того чтобы оценить репродуктивную функцию пациентки, требуется тщательное обследование в клинике ЭКО, включающее в себя исследование матки, эндометрия и овариального запаса яичников.
У пациенток с таким мозаичным вариантом синдрома Шерешевского-Тернера существует некая вероятность развития беременности без применения методов вспомогательной репродукции. При более значительном снижении фертильности могут быть эффективны различные схемы стимуляции овуляции или ЭКО с донорскими ооцитами.
У пациенток с кариотипом 45Х/46XY преодоление бесплодия требует проведения процедуры суррогатного материнства с использованием яйцеклеток донора, т.к. имеет места аплазия матки и влагалища.
Яичники удаляют, поскольку наличие Y-хромосомы в кариотипе повышает риск развития злокачественных процессов.
При таком мозаичном варианте имеет место вирилизация наружных половых органов (увеличенный клитор, измененный вход во влагалище).
Лечение
Лечение различается в каждом конкретном и случае и может предполагать прием анаболических стероидов (для коррекции отставания в росте) и гормональных препаратов (для развития скелета и вторичных половых признаков), а также проведение пластических операций (для удаления крыловидных складок и коррекции косметических дефектов). Помимо этого, проводится терапия выявленных патологий (удвоение почек, дефект межжелудочковой перегородки, остеопороз и т.д.).
Лечение бесплодия при синдроме Шерешевского-Тернера
Схема лечения бесплодия при синдроме Шерешевского-Тернера, как мы уже говорили выше, разрабатывается лечащим врачом гинекологом-репродуктологом в индивидуальном порядке после тщательного обследования.
Хотите записаться на прием?
ПГТ-А: генетическое тестирование на хромосомные аномалии
Диагностика хромосомных аномалий – важнейшая составляющая инновационных репродуктивных технологий. Без преимплантационного генетического тестирования (ПГТ-А) не обходится ни одно планирование беременности в рамках протокола экстракорпорального оплодотворения, что позволяет существенно повысить эффективность самой процедуры.
Суть хромосомного тестирования
ПГТ-А – биопсия клеток наружного слоя бластоцисты – предшественницы эктодермы, которая участвует в получении питательных веществ из матки.
Исследование совершенно безопасно для развития эмбриона, позволяет «увидеть» и выбрать эмбрион без отклонений в хромосомах.
Почему это так важно? Статистика утверждает, что беременность с использованием ЭКО наступает не более чем в 40 % случаев. Причин тому множество, но основной являются хромосомные аномалии.
Чем старше будущая мама, тем выше доля хромосомных отклонений, которые растут в геометрической прогрессии, при этом уверенность в успешной попытке искусственного оплодотворения существенно снижается.
Анеуплоидные зиготы с аномальным числом хромосом встречаются в 70 % случаев.
Именно поэтому проведение генетического тестирования эмбриона еще до его переноса в полость матки – разумно, научно обосновано и несомненно результативно.
Отбор эмбрионов без проведения ПГТ-А только на основании морфологических признаков не может быть надежен на сто процентов. Гарантировать успех в этом случае проблематично. С учетом сложности медицинской процедуры ЭКО, длительности предподготовки к ней, стоимости манипуляций наша клиника и врачи не рекомендуют проводить экстракорпоральное оплодотворение без тестирования ПГТ-А.
Показания к исследованию
Проведение лабораторного исследования на полноценность хромосомного набора рекомендовано в случае:
- постоянного невынашивания беременности (самопроизвольный выкидыш);
- неудачной попытки ЭКО в анамнезе;
- тяжелой патологии сперматогенеза;
- возрастных пациентов (будущие мамы – за 35, мужчины – за 40);
- зафиксированных количественных аномалий хромосом в паре;
- рождения детей с наследственными патологиями в семье;
- скрининга зародышей перед внедрением в матку для повышения эффективности процедуры.
Дополнительно ПГТ-А помогает ответить еще на три важных вопроса:
- каким будет пол ребенка;
- его резус-фактор;
- тканевая совместимость с братьями или сестрами.
Такое исследование проводится по специальным показаниям. Известно, что ряд болезней наследуются исключительно детьми определенного пола.
Так, нарушением свертываемости крови (гемофилией) болеют исключительно представители мужского пола, хотя бессимптомно носить дефектный ген могут мужчины и женщины. Сделав ПГТ-А, можно отобрать только женские эмбрионы.
Это даст 50 % гарантии, что патологический локус просто не передастся девочке по наследству, но все 100% – ее полного здоровья.
Знать резус-фактор будущего малыша необходимо, если папа имеет положительную группу крови, а мама – отрицательную, или наоборот.
Это ситуация с риском резус-конфликта, когда у ребенка и вынашивающей его мамы получается не одинаковый резус-фактор.
Вовремя выявленная хромосомным тестированием аномалия позволит предупредить резус-конфликт, чреватый серьезными осложнениями в период вынашивания плода.
Иногда встает вопрос о необходимости беременности вторым малышом, который будет обладать тканевой совместимостью с уже родившимся братом или сестрой. Тканевую совместимость тоже можно определить с помощью уникального анализа – преимплантационного генетического тестирования.
Ход теста
В процессе планирования беременности в клинике на первом этапе проводят ЭКО или процедуру ИКСИ. Затем сохраняют зиготу в пробирке несколько суток. В лаборатории нашей клиники ПГТ-А тестирование проводится методом Next Generation Sequencing (NGS).
Такое исследование считается максимально достоверным, дает возможность отобрать самый жизнеспособный и здоровый эмбрион для внедрения в матку после тщательного изучения каждой из его нуклеопротеидных структур, хранящих генетическую информацию.
Помимо этого, тест позволяет «увидеть» мозаичный кариотип, несбалансированность хромосомных комбинаций, когда есть лишние хромосомы или их количества недостаточно.
Основное исследование проводится на третьи-пятые сутки развития эмбриона. Для преимплантационного тестирования берут несколько клеток трофэктодермы зародыша, которая трансформируется со временем в плаценту.
Сам эмбрион остается в банке клиники, а биоптат под строжайшим контролем температуры окружающей среды передают в геномную лабораторию.
После анализа результатов с помощью биоинформатики эмбриолог выдает заключение о том, какие эмбрионы могут быть имплантированы.
ПГТ-А исключает риск трансфера генетически неполноценного зародыша, что существенно повышает шансы женщины не только забеременеть, но и выносить, родить здорового ребенка с первой попытки ЭКО.
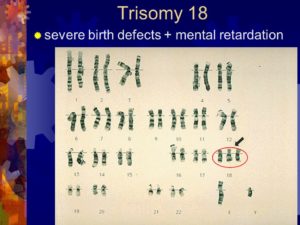
Кариотип характерный для больного с синдромом дауна. Мозаичный синдром дауна. Признаки и симптомы

относится к хромосомным болезням, обусловленным нарушением числа аутосом (неполовых хромосом). (Монголизм) — одна из форм геномной патологии, при которой чаще всего кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями (трисомия).
Тело человека состоит из миллионов клеток, каждая из которых обычно содержит 46 хромосом. Хромосомы расположены парами —половина от матери, половина от отца. У людей с синдромом Дауна в 21-й паре присутствует дополнительная хромосома, вследствие чего, в клетках оказывается по 47 хромосом. При этом у родителей, как правило, нормальный генотип.
В сентябре 2008 года команда ученых из США, Австралии, Испании, Швейцарии и Великобритании прояснила механизм развития синдрома Дауна в эмбриональном периоде .
Как выяснилось, добавочная хромосома повреждает ген REST, который в свою очередь провоцирует целый ряд изменений в генах, регулирующих развитие организма на уровне эмбриональных стволовых клеток.
Запускающим механизмом (триггером) этих изменений является ген DYRK1A, присутствующий в хромосоме 21. Кроме того, тот же участок генома отвечает за развитие болезни Альцгеймера, считают ученые.
Синдромом Дауна называют: синдром трисомии 21, или трисомия по 21 хромосоме.
На электронной микрофотографии показан этот генетический дефект.
Таким образом, существует три формы данного синдрома:Примерно в 95 % случаев возникает ненаследственный вариант болезни — простая полная трисомия 21 хромосомы, обусловленная нерасхождением хромосом во время мейоза. Примерно у 1 % больных наблюдается мозаицизм (не все клетки содержат лишнюю хромосому).
В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21-й хромосомы. Как правило, такие транслокации возникают в результате слияния центромеры 21-й хромосомы и другой акроцентрической хромосомы. Фенотип больных определяется трисомией 21q22.
Повторный риск рождения ребенка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1 % при обычной трисомии у ребенка.
- ненаследственный вариант болезни — 95%
- транслокация хромосомы 21 на другие хромосомы (чаще на 15, реже на 14, ещё реже на 21, 22 и Y-хромосому) — 4 % случаев,
- мозаичный вариант синдрома — 1 %.
ВИДЕО: Как диагностируют синдром Дауна во время беременности
Откуда берется лишняя хромосома
Синдром Дауна получил название в честь английского врача Джона Дауна (John Down), впервые описавшего ее в 1866 году. Связь между происхождением врожденного синдрома и изменением количества хромосом была выявлена только в 1959 году французским генетиком Жеромом Леженом.
Синдром Дауна не является редкой патологией — в среднем наблюдается один случай на 700 родов. Это соотношение одинаково в разных странах, климатических зонах, социальных слоях. Оно не зависит от образа жизни родителей, цвета кожи, национальности.
Ничьей вины в появлении лишней хромосомы нет.
Дополнительная хромосома появляется либо в результате генетической случайности при образовании яйцеклетки или сперматозоида, либо во время первого деления клетки, которое следует за оплодотворением (то есть, когда яйцеклетка и сперматозоид сливаются).
Вероятность рождения детей с синдромом Дауна возрастает с возрастом матери (после 35 лет) и в меньшей мере с возрастом отца. Частота нерасхождения 21-й хромосомы в сперматогенезе, как и в овогенезе, повышается с возрастом.
Для женщин в возрасте до 25 лет вероятность рождения больного ребенка равна 1/1400, до 30 — 1/1000, в 35 лет риск возрастает до 1/350, в 42 года — до 1/60, а в 49 лет — до 1/12.
В данный момент, из-за пренатальной диагностики, частота рождения детей с синдромом Дауна уменьшилась до 1 к 1100.
Соотношение мальчиков и девочек среди новорожденных с синдромом Дауна составляет 1:1.
Тем не менее, поскольку молодые женщины в целом рожают гораздо больше детей, большинство (80%) всех больных синдромом Дауна в действительности рождены молодыми женщинами в возрасте до 30 лет.
А поскольку большинство больных рождается все-таки у молодых матерей, очень важно понять, какие факторы кроме возраста матери влияют на вероятность рождения больного ребенка.
Врачи часто советуют будущим матерям, чей возраст превышает 35 лет, прибегнуть к амниоцентезу, т.е. процедуре получения образца околоплодных вод для анализа хромосомного состава клеток.
Это дает возможность прервать беременность, угрожающую рождением больного ребенка.
Генетическая вероятность появдения ребенка с синдромом Дауна
Недавно индийские ученые обнаружили, что вероятность рождения ребенка с синдромом Дауна сильно зависит от возраста бабушки по материнской линии: чем старше она была, когда рожала дочь, тем выше вероятность рождения больных внуков.
Этот фактор может оказаться более значимым, чем три других, известных ранее (возраст матери, возраст отца и степень близкородственности брака). Malini S. S., Ramachandra N. B.
Influence of advanced age of maternal grandmothers on Down syndrome// BMC Medical Genetics. 2006, 7:4.
Слово «синдром» означает набор признаков или характерных черт.
В 1866 году в своей первой статье Дж. Лэнгдон Даун описал некоторые характерные черты людей с синдромом Дауна. Он отметил, в частности, такие специфические особенности лица как: плоский профиль, узкие, широко расставленные раскосые глаза.
Обычно синдрому Дауна сопутствуют следующие внешние признаки:
- «плоское лицо» — 90%
- монголоидный разрез глаз — 80%
- брахицефалия (аномальное укорочение черепа) — 81%
- плоский затылок — 78%
- плоская переносица — 52%
- короткий нос — 40%
- кожная складка на шее у новорожденных — 81%
- короткая широкая шея — 45%
- Мочки ушей плохо развиты и оказываются приросшими.
- эпикант (вертикальная кожная складка, прикрывающая медиальный угол глазной щели) — 80%
- гиперподвижность суставов — 80%
- мышечная гипотония — 80%
- катаракта в возрасте старше 8 лет — 66%
- страбизм = косоглазие — 29%
- пигментные пятна по краю радужки = пятна Брушфильда — 19%
- открытый рот (в связи с низким тонусом мышц и особым строением нёба) — 65%
- аркообразное («готическое») нёбо — 58%
- бороздчатый язык — 50%
- зубные аномалии — 65%
- короткие конечности — 70%
- брахимезофалангия (укорочение всех пальцев за счет недоразвития средних фаланг) — 70%
- клинодактилия 5-го пальца (искривленный мизинец) — 60%
- поперечная ладонная складка (называемая также «обезьяньей») — 45%
- ВПС (врожденный порок сердца) — 40%
- деформация грудной клетки, килевидная или воронкообразная, — 27%
- эписиндром — 8%
- Аномалии ЖКТ — 10-18%
- стеноз или атрезия 12-перстной кишки — 8%
- врожденный лейкоз — 8%.
Больные с синдромом Дауна имеют низкий рост, хриплый голос, умственную отсталость (типичный IQ между 30 і 50).
Врожденные пороки сердца являются характерными признаками синдрома Дауна. Они встречаются у 40% больных. Чаще всего это: атриовентрикулярная коммуникация и дефекты межжелудочковой перегородки .
Для синдрома Дауна характерна поперечная ладонная складка (называемая также «обезьяньей»).
Большинство мужчин с синдромом Дауна бесплодны, а 50 % женщин с синдромом Дауна могут иметь детей.
35-50 % детей, рожденных от матерей с синдромом Дауна, рождаются с синдромом Дауна или другими отклонениями. Интересно, что больные синдромом Дауна реже имеют раковые опухоли.
Видимо, 21-ая хромосома содержит ген-«глушитель опухолей», и наличие третьей копии гена обеспечивает дополнительную защиту против рака.
Установлено, что если синдромом Дауна страдает один из однояйцовых близнецов, то неизбежно болен и другой, а у разнояйцовых близнецов, как и вообще у братьев и сестер, вероятность такого совпадения значительного ниже.
Данный факт дополнительно свидетельствует в пользу хромосомного происхождения болезни.
Однако синдром Дауна нельзя считать наследственным заболеванием, так как при нем не происходит передачи дефектного гена из поколения в поколение, а расстройство возникает на уровне репродуктивного процесса.
Точная диагностика
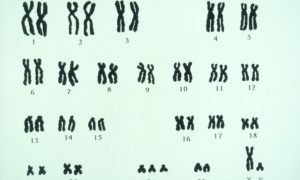
Диагностировать синдром Дауна безошибочно возможно на основании анализа крови на кариотип.
(Анализ показывает набор хромосом у каждого из супругов, необходим для выявления хромосомных заболеваний. Например, аномальный кариотип человека для синдрома Дауна, Трисомия по 21-й хромосоме: 47, ХХ, 21+; 47, ХY, 21+).
| Кариотипы | Болезнь |
Моносомия хромосомы Х: что это такое, возможно ли лечение отклонения и беременность при таком диагнозе
Хромосомы, отвечающие за пол, обладают особенным свойством. Если нарушения в прочих частях ядра чреваты внутриутробной гибелью плода или тяжелыми патологиями, то при изменении количества половых хромосом возможно рождение вполне жизнеспособного ребенка.
В зависимости от вариации набора различается и дальнейшее развитие. Например, многие обладательницы вида моносомии — трисомии по Х-хромосоме или носители кариотипа XYY даже не догадываются о своей необычности.
В других случаях улучшить качество жизни помогает современная терапия.
Девочкам с единственной Х-хромосомой, несмотря на их особенность, медицина тоже дает шанс преодолеть все трудности, связанные с синдромом моносомии. Но некоторые ограничения в настоящий момент по-прежнему непреодолимы.
Моносомия хромосомы Х: понятие
Моносомия X-хромосомы — генетическое нарушение, также известное как синдром Шерешевского-Тернера. Такое название оно получило по фамилиям исследователей, впервые давших точное описание этой патологии в начале ХХ века.
Советский доктор наук Н.А. Шерешевский занимался эндокринологией и считал это состояние болезнью, вызываемой недостаточным развитием половых желез и гипофиза.
Но в 1959 году британский генетик Чарльз Форд выявил хромосомную природу происхождения.
В зарубежной литературе иногда используют термин «синдром Ульриха-Тернера» в честь еще одного ученого, занимавшегося исследованием этой проблемы.
Люди с синдромом Шерешевского-Тернера (СШТ) обладают набором из 45 хромосом вместо обычных 46.
Их кариотип выглядит как 45 X0 и носит в какой-то степени уникальный характер. Никакая другая моносомия не дает жизнеспособный человеческий организм, способный к относительно стабильному развитию.
Все эмбрионы с нехваткой аутосомных хромосом или кариотипом Y0 гибнут в первые месяцы внутриутробного периода.
Разновидность моносомиии
Как и при других генетических патологиях, эта мутация может иметь мозаичную форму. Более того, ее уровень является одним из самых высоких среди других генетических нарушений — около 40%.
В этом случае внешние проявления могут быть значительно сглажены или практически отсутствовать, в зависимости от того, какой объем клеток в организме имеет аномальный хромосомный набор.
Некоторые из носителей узнают о своей особенности только после кариотипирования при проблемах с зачатием.
Существуют следующие варианты мозаицизма:
- кариотип 45X/46XX — в организме присутствуют клетки сразу с нормальным и аномальным женским набором;
- кариотип 46 Х Хр- и 46 X Xq— — отсутствие части хромосомы или так называемая делеция плеч;
- кариотип 46 Xi(Xq) или 46, X, i(Xp) — наличие изохромосомы с идентичными плечами;
- кариотип 45 Х/46 XY — в организме присутствует мужская половая хромосома, способная дать частичное развитие соответствующего типа, но в большинстве случаев гениталии амбисексуальны — в брюшной полости одновременно находится неопустившееся яичко и зачаточная матка.
По каким причинам происходит патология
Причина моносомии кроется в мутации, которая возникает спонтанно при образовании плодного яйца в первые дни беременности. Одна из родительских Х-хромосом (в большинстве случаев отцовская) частично повреждается или вовсе выпадает из процесса формирования кариотипа.
В отличие от многих других генетических аномалий, данное нарушение не зависит от возраста родителей.
Протекание беременности при СШТ у плода почти всегда сопровождается угрозой прерывания. Возможны преждевременные роды.
Методы диагностики
СШТ можно выявить пренатально, но в большинстве случаев нарушение диагностируется уже после рождения ребенка. Во время беременности материалом для исследования служат ворсины хориона, околоплодная жидкость или пуповинная кровь. У появившихся на свет девочек для генетического анализа берут кровь из вены.
Важно знать всем женщинам: что такое аденоз молочных желез, симптомы и методы лечения заболевания.
Из-за чего возникает гематометра, и каких осложнений опасаться читайте тут.
О прыщах перед месячными: https://venerolog-ginekolog.ru/gynecology/diseases/pryishhi-pered-mesyachnyimi.html.
Лечение и наблюдение пациентов
Полностью ликвидировать проявления синдрома нельзя, но качество жизни улучшает гормональная терапия.
Начиная с младшего школьного возраста девочкам показан прием соматотропина для приближения конечного роста к норме.
В подростковом возрасте применение гормональных препаратов восполняет недостаток собственного эстрогена, что помогает организму сформироваться по женскому типу — как внешне, так и внутренне.
Вероятность здоровой беременности
Несмотря на то, что заместительная гормональная терапия позволяет вырастить репродуктивную систему до взрослого состояния, истинный СШТ, как правило, сопровождается полным отсутствием собственных яйцеклеток.
Однако при мозаичной разновидности патологии есть вероятность их созревания, нарушения гормонального фона проявляются в нестабильном менструальном цикле. Естественная беременность и успешные роды наступают в единичных случаях, чаще всего в этот период требуется гормональная поддержка для предотвращения выкидышей.
В большинстве же случаев необходимо использование процедуры экстракорпорального оплодотворения, иногда — с донорской яйцеклеткой.
Если обладательнице мозаичного СШТ удалось забеременеть, вероятность родить здорового ребенка у нее не намного ниже, чем у обычных женщин.
Интересное видео о X-хромосоме:
Мозаичная форма синдрома Шерешевского-Тернера

Что это такое, синдром Шерешевского-Тернера? Этот вопрос нередко интересует людей, столкнувшимся с этим заболеванием.
Данная патология являет собой врожденную хромосомную аномалию, которая характеризуется отсутствием одной X-хромосомы (классический вариант).
Учитывая особенности кариотипа, который записывается как 45 XO, очевидным является тот факт, что страдают от соответствующей патологии только представительницы женского пола.
Синдром Шерешевского-Тернера мозаичная форма – патология, при которой не обязательно полностью отсутствует вторая X-хромосома. Чаще она имеет некоторые дефекты, влияющие на общее состояние здоровья пациентки и протекание ее заболевания.
Причины
Причины синдрома Шерешевского-Тернера сокрыты в нарушениях деления клеток и образования генетического материала ребенка еще в период зачатия. Патология фактически возникает в самом начале развития плода.
Нехватка или неправильное распределение целых молекул ДНК ведет к нарушению структуры отдельных хромосом. В данном случае страдает X-хромосома.
При классической форме заболевания она просто отсутствует – 45 XO (в норме 46 XX).
Нехватка генетического материала является причиной развития целого ряда характерных симптомов, которые существенно отражаются на качестве жизни девочки уже с самого рождения.
Однако стоит отметить, что в отличие от некоторых хромосомных аномалий при синдроме Шерешевского-Тернера сохраняется жизнеспособность, а женщины могут вести полноценную жизнь, беременеть и рожать детей.
Факторами, которые способствуют возникновению хромосомной аномалии, являются:
- Половые инфекции во время беременности женщины или в анамнезе.
- Влияние некоторых физических факторов (радиация, электромагнитные поля).
- Влияние химикатов на плод.
- Наследственный фактор.
- Истощение и голодание в период вынашивания плода.
Заболевание может развиваться, даже если перечисленные выше факторы отсутствуют.
Что такое мозаицизм?
Синдром Шерешевского-Тернера мозаичная форма считается более благоприятным заболеванием по сравнению с классической формой, и протекает легче обычного. Причины его возникновения те же, что и указаны выше.
Главным отличием от классической формы болезни является частичное развитие патологии в клетках. В организме одновременно существуют ткани с нормальным кариотипом и аномальным.
Это позволяет компенсировать проявление некоторых симптомов, что делает течение болезни более легким.
В зависимости от характера повреждения второй X-хромосомы, выделяют следующие варианты мозаичной формы заболевания (по кариотипу):
- 46 Х Хр и 46 X Xq.
- 46 Xi(Xq) или 46,X,i(Xp).
- 45 Х/46 XY.
Как видно, практически во всех случаях общее количество хромосом остается нормальным (46). Но, несмотря на этот факт, присутствуют определенные дефекты в плечах одной из них.
Именно данная ситуация является причиной возникновения характерных признаков и симптомов.
Они вызваны преимущественно изменениями в процессе синтеза белков, что, в конечном счете, проявляется различными аномалиями развития.
Клиническая картина
Симптомы синдрома Шерешевского-Тернера достаточно специфичны. Во многих случаях диагноз удается установить визуально при рождении ребенка. Наиболее распространенными признаками патологии являются:
- Недоразвитие половой системы. Оно проявляется отсутствием или слишком маленьким размером яичников, гипоплазией матки и фаллопиевых труб. В пубертатном возрасте нормально не развиваются молочные железы. Соски часто втянуты, широко расположены.
Наблюдается снижение нормальных концентраций половых гормонов в крови, что является основной причиной нарушений менструального цикла. Он может вовсе отсутствовать или быть нерегулярным со скудными выделениями. Во многих случаях естественная беременность невозможна.
- Аномалии развития внутренних органов. Данная группа симптомов чаще проявляется стенозом отверстий между камерами сердца, гипоплазией или патологическими изменением структуры почек.
- Другие симптомы. Отмечается низкий рост. Частым признаком болезни является «шея сфинкса» – формирование кожных складок в данной области. Отеки лица, вальгусное искривление локтей, эпикант и деформация грудной клетки также могут выступать признаками синдрома Шерешевского-Тернера.
Учитывая полиморфизм проявлений классической патологии, постановка диагноза редко бывает проблематичной.
Мозаичная форма синдрома Шерешевского-Тернера отмечается более легким протеканием заболевания. Фенотип (внешние проявления) может оставаться нормальным. Единственным обязательным симптомом практически у всех пациенток остается патология развития половой системы. Степень выраженности заболевания зависит от количества пораженных клеток в организме.
Диагностика
Диагностика синдрома Шерешевского-Тернера в большинстве случаев не составляет особого труда, если речь идет о классической форме болезни. Что же касается мозаичной формы патологии, то здесь иногда возникают отдельные трудности. Дело в том, что при нормальном фенотипе клиницисты далеко не всегда могут сразу заподозрить хромосомную патологию.
Лучшим способом диагностики болезни остается кариотипирование. Оно предусматривает анализ хромосомного набора пациентки с установлением наличия дефектов или отсутствия X-хромосомы.
Для верификации болезни у плода проводят пренатальную диагностику. Главными ее этапами являются:
- Выявления потенциальных факторов риска рождения больного ребенка.
- Анализ кариотипа родителей.
- УЗИ плода.
- Кариотипирование малыша еще в утробе.
С помощью этих методов можно установить наличие болезни еще до момента рождения ребенка. Это позволит сразу же начать соответствующую терапию с улучшением качества жизни девочки в будущем.
Осложнения
Осложнения при мозаичной форме синдрома Шерешевского-Тернера в основном связаны с наличием аномалий развития внутренних органов. Пороки сердца могут привести к ранним проблемам с кровообращением. Недоразвитие почек является причиной почечной недостаточности. Расщепление позвоночника приводит к неврологической симптоматике и тому подобное. Все зависит от исходного состояния организма.
Мозаичная форма болезни в этом плане намного благоприятнее. Опасные осложнения практически не возникают. Патология ограничивается снижением качества жизни, что не слишком влияет на ее продолжительность.
")
")



