Синдром делеции 22 хромосомы (синдром ДиДжорджи) у детей. Клинические рекомендации

- Аутоиммунные осложнения
- Велофарингеальная недостаточность
- Врожденный порок сердца
- Гипокальциемия
- Гипопаратиреоз
- Делеция 22 хромосомы
- Дефект Т-клеточного звена
- Задержка речевого и психомоторного развития
- Задержка физического развития
- Внутривенные иммуноглобулины
- Инфекционные осложнения
- Расщепление неба и верхней губы
- Синдром ДиДжорджи
АИГА – аутоиммунная гемолитическая анемия
АЛТ — аланинаминотрансфераза
АСТ — аспартатаминотрансфераза
ВВИГ — внутривенные иммуноглобулины
ВЗК – воспалительные заболевания кишечника
ВПС – врожденный порок сердца
ГКС — глюкокортикостероиды
ДНК — дезоксирибонуклеиновая кислота
ЖКТ — желудочно-кишечный тракт
ИТП – иммунная тромбоцитопения
КМ — костный мозг
КТ — компьютерная томография
ЛОР — ларингооторинолог
ЛПУ — лечебно-профилактическое учреждение
МЗ — Министерство здравоохранения
МКБ-10 — Международная классификация болезней 10-го пересмотра
МРТ —магнитно-резонансная томография
РКИ — рандомизированные контролируемые исследования
РНК — рибонуклеиновая кислота
РФ — Российская Федерация
Синдром del 22q11 – синдром делеции 22 хромосомы=синдром ДиДжорджи
СДД — синдром ДиДжорджи
ТГСК – трансплантация гемопоэтических стволовых клеток
УЗИ – ультразвуковое исследование
ЭКГ – электрокардиограмма
ЭхоКГ -эхокардиография
ЮРА – ювенильный ревматоидный артрит
del 22q11.2 – делеция длинного плеча 22 хромосомы локус 11.2
САТСН 22 — Cardiac defects, Abnormal facies, Thymic hypoplasia, Cleft palate, Hypocalcemia, 22q deletion – порок сердца, лицевые аномалии, гипоплазия тимуса, расщелина неба, гипокальцемия, делеция 22
FISH –fluorescent in situ hybridization — флуоресцентная гибридизация in situ
ТВХ 1 ген –Т бокс 1 ген
TREC — T-cell Receptor Excision Circles
Термины и определения
Внутривенные иммуноглобулины – препараты, содержащие преимущественно нормальный человеческий IgG. Изготовляются из пулированной плазмы тысяч здоровых доноров, с применением специальных методов очистки и вирусинактивации.
Делеция – потеря участка хромосомы
Хромосомные аберрации – изменение числа иили структуры хромосом
Микрогнатия — недоразвитие (гипоплазия) челюстных костей.
Ретрогнатия — смещение челюстной кости в дорзальном направлении (кзади)
Гипертелоризм — увеличенное расстояние между глазами
TREC – кольцевые фрагменты ДНК, образующиеся при развитии Т лимфоцитов в тимусе, в частности, в процессе формирования Т клеточного рецептора. Их концентрация в крови отражает эффективность тимопоэза. Используется для скрининга Т клеточных иммунодефицитов.
Трансплантация гематопоэтических стволовых клеток – метод лечения некоторых наследственных и приобретенных гематологических, онкологических и иммунных заболеваний, основанный на замене собственного, патологического кроветворения больного на нормальное кроветворение донора.
1.1 Определение
Синдром делеции 22-й хромосомы (синдром del 22q11) или синдром ДиДжоржи (СДД) — это совокупность морфологических, иммунологических и неврологических изменений, которые являются следствием делеции длинного плеча одной копии 22-й хромосомы — del 22q11.2 [1,2].
В классическом понятии этот синдром представляет собой комплекс симптомов, состоящий из патологии лицевого скелета (расщелины твердого неба), врожденного порока сердца, иммунодефицита вследствие гипоплазии (аплазии) тимуса и гипокальциемии, как результат гипоплазии паращитовидной железы [1,3].
Как ни один другой синдром, синдром del 22q11.2 вариабелен в количестве признаков и степени их выраженности, что и объясняет тот факт, что этот синдром в литературе имеет порядка десятка различных названий, включая синдром ДиДжорджи, САТСН 22, велокардиофациальный сидром, Шпринтцена синдром, Кайлера синдром, синдром лицевых и конотрункальных аномалий и т.д.[1,3,4].
1.2 Этиология и патогенез
В основе заболевания лежит нарушение формирования органов, происходящих их третьей жаберной дуги (нижняя часть лицевого скелета, тимус, паращитовидная железа, верхние отделы сердца и магистральных сосудов). Цитогенетические и молекулярные исследования показали, что делеция 22q11.
2 является ведущей причиной СДД и возникает спорадически более чем в 90% случаев [5,6,7]. В 10% случаев делеция наследуется от одного из родителей, так как наследование происходит аутосомно- доминантным путем [1,4].
В редких случаях синдром является проявлением перестроек других хромосом, а также мутации гена ТВХ1 [4].
Анализ ДНК пациентов с СДД хромосомы выявил, что в 85-90% случаев выпадающий участок является одним и тем же. Дефект находится между D22S427 на 22q11.21 и D22S801 на 22q11.23. В этом участке локализовано не менее 40 генов, что составляет около 3 млн пар нуклеиновых оснований.
В 10-12% случаев встречаются более короткие делеции, которые составляют 1,5-2 млн парных оснований. Было описано несколько пациентов с синдромом делеции 22-й хромосомы, имеющих делеции за пределами наиболее часто выпадающих участков [5,6].
Результаты проведенных исследований свидетельствуют о том, что степень выраженности фенотипа не коррелирует с размером делеции, т.е. пациент с потерей 1,5 млн парных оснований может иметь такой же по тяжести фенотип, как и с делецией в 3 млн парных оснований [2,5].
Кроме того, было замечено, что вариабельность фенотипических проявлений варьирует как внутри одной семьи, так и между семьями, несмотря на идентичные участки делеции [5].
Делеция вызывает выпадение участка, включающего ген ТВХ, ген фактора транскрипции, участвующего в развитии фарингеальных дуг [5,6]. Эти изменения, в свою очередь, ведут к нарушению формирования сердца и магистральных сосудов, иммунологическим изменениям, расщеплению нёба и верхней губы, гипопаратиреоидизму, задержке умственного развития.
Несмотря на то, что ТВХ1, без сомнения, является главным геном, формирующим фенотип при синдроме делеции 22-й хромосомы, в результате исследований были выявлены и другие гены, недостаточная экспрессия которых может играть роль в формировании фенотипических проявлений [6,7].
Учитывая результаты работ по выяснению молекулярных основ заболевания, ясно, что в формировании фенотипа играет роль комплексное нарушение экспрессии и взаимодействия генов, их модификаторов и других составляющих, что приводит к дальнейшему нарушению эмбрио- и органогенеза [4,5,7].
Соответственно, при отсутствии или нарушении функции и экспрессии генов и дальнейших процессов происходит формирование пороков развития, характерных для СДД [1,5].
1.3 Эпидемиология
Синдром делеции 22-й хромосомы — одна из самых частых делеций среди других хромосомных аберраций в человеческом геноме, по частоте она уступает лишь синдрому Дауна, трисомии по 21-й хромосоме. Частота встречаемости варьирует от 1:4000 до 1:6000 новорожденных [1,2,3].
Не наблюдается ни половой, ни этнической предрасположенности к данному синдрому. Большинство пациентов с СДД имеют патологию лицевого скелета и врожденный порок сердца и развивают гипокальциемию вскоре после рождения [6].
Пациенты, не имеющие данных симптомов, зачастую диагностируются в раннем возрасте, и правильный значительно запаздывает.
1.4 Кодирование по МКБ-10
Другие иммунодефициты (D84):
D84.1 – Синдром ДиГеорга.
1.5 Классификация
Исторически сложилось, что в литературе часто используется разделение синдрома на полный и неполный (частичный) [1,3,5]:
- Термин «Полный синдром ДиДжорджи» использовался у пациентов, имеющих полный спектр типичных проявлений, включая выраженный иммунодефицит.
- Термин «Частичный синдром ДиДжорджи» использовался у пациентов, если они имели лишь некоторые типичные признаки, особенно без проявлений выраженного иммунодефицита. Частичный синдром делеции 22-й хромосомы в значительной степени превалирует по количеству в сравнении с полным.
2.1 Жалобы и анамнез
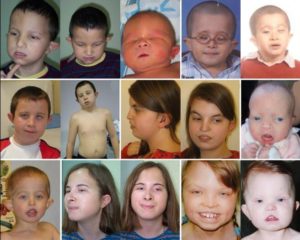
Спектр клинических проявлений при синдроме делеции 22-й хромосомы достаточно широк [3,6,9,11,12,14,15], поэтому жалобы и анамнез заболевания могут быть крайне разнообразными и различными по степени выраженности:
- Врожденный порок сердца представлен не менее, чем в 80% случаев. Некоторые из пороков являются более патогномоничными: прерывание дуги аорты, общий артериальный ствол и тетрада Фалло являются наиболее частыми среди данной группы детей [6,11].
- Гипокальциемия/гипопаратиреоз может проявляться судорожным синдромом при выраженном дефиците кальция в младенческом возрасте [6,12].
- Поражение носоглоточного аппарата выявлено примерно в 70% случаев и проявляется в виде велофарингеальной недостаточности, расщеплении нёба, губы, раздвоении уздечки нёба, гнусавым оттенком голоса, также описано снижение обоняния, кондуктивная и/или сен- соневральная тугоухость [6,10,13].
- Характерные черты лица (удлиненное лицо, микрогнатия или ретрогнатия, широкая переносица, мелкие зубы, асимметрия лица при плаче, опущенные вниз уголки рта, глазной гипертелоризм, низко посаженные и деформированные ушные раковины, бульбообразный кончик носа) [2,5,10].
- Иммунологические нарушения встречаются в 77% случаев. Однако инфекционные проявления вследствие дефекта иммунной системы дебютируют не с рождения. Чаще других звеньев поражается Т-клеточное звено, что проявляется предрасположенностью к грибковым заболеваниям, пневмоцистной инфекции, некоторым бактериальным и вирусным инфекциям [1,8,10].
- Нарушение выработки Т-клеток может предрасполагать к аутоиммунным заболеваниям. Описаны такие осложнения синдрома делеции 22-й хромосомы, как ЮРА, ХТП, АИГА, ВЗК, болезнь Грейвса, аутоиммунный увеит, бронхиальная астма [1,9,10,14].
- Задержка физического развития нередко наблюдается у пациентов с синдромом делеции 22q11.2-й хромосомы, которые несколько отличаются от стандартных таблиц [1,2,6,10].
- Задержка речевого и психомоторного развития наблюдается у 70—90% и проявляется с возрастом, однако тестирование пациентов с задержкой развития имеет смысл только в случаях сочетании с другими признаками [2,10,15].
2.2 Физикальное обследование
Физическое развитие большинства пациентов низкое и дисгармоничное по весу [1,10].
Стигмы дисэмбриогенеза широко вариабельны и не являются патогномоничными, однако чаще других признаков обращают на себя внимание глазной гипертелоризм, бульбообразный кончик носа и низко посаженные и или деформированные ушные раковины [6,10,13].
Могут проявляться признаки дыхательной и сердечной недостаточности [2,11]. Могут встречаться пороки развития дыхательной, пищеварительной, костно-мышечной и других систем.
Задержка умственного и речевого развития встречается у подавляющего числа пациентов с данным синдромом [2,3,15].
2.3 Лабораторная диагностика
- Рекомендуется клинический анализ крови [2,3].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 3)
- Рекомендуется исследование уровня ионизированного кальция, паратиреоиного гормона [12].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2)
- Рекомендуется исследование уровня гормонов щитовидной железы [12].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 3)
- Рекомендуется исследование уровня сывороточных иммуноглобулинов и клеточного иммунитета, включая определение количества наивных Т-лимфоцитов [1,10,16,17].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2)
Субтеломерные делеции 1q43q44 и тяжелые поражения головного мозга, связанные с задержкой миелинизации | журнал генетики человека

- перегласовка
- Миелин биология и ремонт
- Нарушения развития нервной системы
Субтеломерные делеции 1q44 вызывают задержку умственного развития, задержку развития и аномалии головного мозга, в том числе аномалии мозолистого тела (ACC) и микроцефалию у большинства пациентов.
Мы сообщаем о случаях шести пациентов с делециями 1q44; два пациента с интерстициальными делециями 1q44; и четыре пациента с терминальными делециями 1q. У одного из пациентов наблюдалась несбалансированная транслокация между хромосомой 5.
Все области делеции перекрывались с ранее сообщенными критическими областями для ACC, микроцефалии и судорог, что указывает на рецидивную природу основных фенотипических признаков делеций 1q44.
У четырех пациентов с терминальными делециями 1q обнаружена сильная потеря объема в мозге по сравнению с пациентами, у которых были интерстициальные делеции 1q44. Это указывало на то, что теломерные области играют важную роль в серьезной потере объема мозга.
Кроме того, у двух пациентов с терминальными делециями 1q43, за пределами критической области для синдрома делеции 1q44, наблюдалась отсроченная миелинизация. Поскольку области делеции, идентифицированные у этих пациентов, расширяются к центромере, мы заключаем, что гены, ответственные за задержанную миелинизацию, могут быть расположены в соседней области 1q43.
Субмикроскопические субтеломерные хромосомные делеции были обнаружены у 7, 4% детей с умственной отсталостью от умеренной до тяжелой степени. 1 Некоторые субтеломерные делеционные синдромы являются клинически узнаваемыми и идентифицируются по характерным признакам, тогда как некоторые другие не могут быть идентифицированы с помощью таких средств.
Недавнее развитие молекулярного кариотипирования с использованием хромосомных микрочипов выявило четкие корреляции генотип-фенотип и выявило критические хромосомные области с характерными признаками субтеломерных делеций. Наиболее ярким примером является синдром Миллера-Дикера, который показал четкие корреляции генотип-фенотип.
2 Синдром Миллера-Дикера вызывается субтеломерной делецией 17p и хорошо распознается и характеризуется лиссэнцефалией и отличительными чертами лица 3, которые являются результатом участия гена регуляторной субъединицы 1 фактора активации тромбоцитов 1b ( PAFAH1B1 ) и тирозина Ген 3-монооксигеназы / триптофана 5-монооксигеназы-активирующий эпсилон-полипептидный ген ( YWHAE ) соответственно; оба эти гена расположены на 17p13. 3
Несколько исследований исследовали критическую область для синдрома субтеломерной делеции 1q44 и обнаружили, что основными фенотипическими признаками синдрома субтеломерной делеции 1q44 являются микроцефалия, аномалии мозолистого тела (ACC) и судороги. 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 Недавно Ballif et al. 17 проанализировали пациентов с микроделециями 1q44 и предложили определенные гены, которые могут отвечать за индивидуальные особенности.
Мы сообщаем о случаях шести вновь выявленных пациентов с делециями 1q44; два с интерстициальной делецией 1q44; и четыре с удалением терминала 1q. Поскольку у пациентов с терминальной делецией 1q44 обнаружены более тяжелые фенотипы по сравнению с пациентами с интерстициальной делецией, фенотипические различия будут получены из дополнительно удаленной области 1q43q44.
Хромосомные делеции
Тестирование хромосомных микрочипов выявило аберрации в области 1q43q44 у шести пациентов (рис. 1). У пациентов 1 и 2 были удалены интерстициальные делеции 1, 9 и 2, 2 Мб, соответственно, в области 1q43q44. У пациента 1 анализ FISH подтвердил удаление (рис. 2а).
Последующий анализ FISH не выявил аномалий у родителей обеих семей, что указывает на делеции de novo у обоих пациентов.
Молекулярное кариотипирование определяло аберрации как arr 1q44 (243 809 193–245 665 521) × 1 дн для пациента 1 и arr 1q43q44 (243 303 991–245 506 920) × 1 дн для пациента 2.
Результаты тестирования хромосомных микрочипов, представленные в Gene View Agilent Genomic Workbench (Agilent Technologies). Вертикальная ось и горизонтальная ось представляют отношение сигналов log 2 и геномное положение соответственно. Аберрантные области показаны синими прямоугольниками. Точки указывают положения генома и отношение log 2 каждого зонда.
Изображение в полном размере
Результаты анализов FISH. ( a ) Потеря зеленой маркировки сигнала RP11-7L23 (стрелка) указывает на удаление 1q44 у пациента 1. ( b ) Потеря зеленой маркировки сигнала RP11-88N11 (стрелка) указывает на удаление 1q44 у пациента 3.
( c — e ) Подтверждение несбалансированной транслокации между хромосомой 1 и 5 у пациента 4. Потеря зеленой маркировки сигнала RP11-143E8 (стрелка) указывает на делецию 1q44 ( c ). Дополнительная сигнальная метка RP11-94J21 5p15.
33 присутствует на другой хромосоме ( d, красный сигнал, стрелка), что указывает на транслокацию на хромосому 1 ( e, зеленый сигнал, стрелка).
Изображение в полном размере
У пациента 3 терминальные делеции 1q43 были идентифицированы и подтверждены с помощью анализа FISH (рис. 2b). Поскольку анализ FISH для обоих родителей не выявил никаких отклонений, эта делеция произошла как de novo . Молекулярное кариотипирование пациента 3 было обозначено как обр 1q43q44 (242 442 098–249 250 621) × 1 дн.
У пациента 4 была выявлена потеря числа копий генома в 1q43q44 и дополнительное усиление в 5p15.33 (дополнительная фигура 1). Последующий анализ FISH подтвердил несбалансированную транслокацию между 1q43 и 5p у пациента 4 (Рисунки 2c-e), и ни у одного из родителей транслокация не была обнаружена.
Следовательно, было установлено, что транслокация дисбаланса у пациента была de novo по происхождению. Ее кариотип был 46, XX, дер (1) т (1; 5) (q43; с15, 33) .arr 1q43q44 (242 223 230–249 212 668) × 1, 5p15, 33 (57 640–1 705 515) × 3 дн.
Дублированная область 5p была только 1, 7 Мб из терминальной области.
У пациента 5 терминальные делеции 1q43 были идентифицированы с точкой разрыва в гене гомолога 3 вирусного онкогена v-akt мышиной тимомы ( AKT3 ). Молекулярный кариотип был arr 1q44 (243 880 099–249 212 668) × 1. Наибольшая делеция была выявлена у пациента 6 с arr 1q43q44 (238 888 870–249 212 668) × 1.
Результаты анализа FISH приведены в дополнительной таблице 1.
Клинический отчет
9-летний мальчик родился вакуумной экстракцией. Его масса тела при рождении составляла 2820 г (в пределах 25-го центиля), длина составляла 45 см (= 3-й центиль), а окружность затылочно-лобного отдела тела (OFC) составляла 32, 5 см (10-25-й центиль).
После рождения у него появились проблемы с кормлением. Его развитие было слегка задержано: контроль над головой был достигнут через 8 месяцев, сидя без поддержки в 15 месяцев, ползая через 24 месяца и ходя в одиночестве без поддержки через 44 месяца.
В возрасте 11 месяцев он перенес рецидив фебрильных судорог. В 18 месяцев он перенес нефебрильные судороги.
Магнитно-резонансная томография головного мозга (МРТ) в возрасте 4 лет показала потерю объема в лобной доле, легкие аномальные формы гираля в лобной доле и боковой доле, а также ACC (Рисунки 3a – c). Обычный хромосомный анализ выявил нормальный мужской кариотип.
Результаты МРТ головного мозга пациента 1, осмотренного в 4 года ( a — c ), пациента 2, осмотренного в 3 года ( d — f ), пациента 3, осмотренного в 12 месяцев ( g — i ), пациента 4, осмотренного в 15 месяцев ( j — 1 ) пациент 5 обследован через 5 месяцев ( м- о ), а пациент 6 обследован через 3 года ( р- р ). T1-взвешенные сагиттальные виды ( a, d, g, j, m, p ), T1-взвешенные осевые виды ( b, e, h, k, n, q ) и T2-взвешенные осевые виды ( c, f, i, l, o, r ). АКК отмечены у всех пациентов. Пациент 5 ( м ), в частности, демонстрирует полный агенез мозолистого тела. Уменьшение объема лобной доли наблюдается у всех пациентов. Заметно отсроченная миелинизация отмечена у пациентов 3 и 4 ( i и l ).
Изображение в полном размере
В настоящее время его рост составляет 119, 5 см (
Хромосомные патологии при беременности
:
Хромосомные патологии при беременности, к сожалению, совсем не такая редкость, как хотелось бы.
Но, есть и хорошая новость – многие хромосомные патологии, по крайней мере, самые часто встречающиеся, можно диагностировать с помощью различных тестов еще на ранних сроках беременности.
Конечно же, вылечить это невозможно даже при самой ранней диагностике, но у будущих родителей хотя бы появляется выбор – готовы ли они к рождению особенного ребенка, или примут решение прервать беременность.
Если родители в любом случае решают оставить малыша, какое заболевание не было бы диагностировано, эта информация дает возможность максимально подготовиться к жизни после родов, обдумать, как и где они смогут лечить ребенка, будут теоретически знать, какие особенности ухода нужно иметь в виду и прочие важные нюансы.
Для начала посмотрим, что такое хромосомные патологии, какие они бывают, и что такое вообще хромосомы.
Хромосомы представляют собой палочковидные структуры в середине каждой клетки в организме. Каждая клетка имеет 46 хромосом, сгруппированных в 23 пары. Когда хромосома является ненормальной, это может вызвать проблемы со здоровьем в организме. Аномальные хромосомы чаще всего образуются в результате ошибки во время деления клетки.
Аномалии хромосом часто происходят из-за одного или нескольких факторов:
- Ошибки при делении половых клеток (мейоз)
- Ошибки при делении других клеток (митоз)
- Воздействие веществ, вызывающих врожденные дефекты (тератогены)
Мейоз
Мейоз — это процесс, при котором половые клетки делятся и создают новые половые клетки с половиной числа хромосом. Сперматозоиды и яйцеклетки — это половые клетки. Мейоз — это начало процесса развития ребенка. Когда сперматозоид оплодотворяет яйцеклетку, соединение приводит к зачатию ребенку с 46 хромосомами – это в случае нормы.
Но если мейоз не происходит нормально, у ребенка может быть лишняя хромосома (трисомия) или недостающая хромосома (моносомия). Эти проблемы могут привести к потере беременности, или они могут вызвать проблемы со здоровьем у ребенка.
Женщина в возрасте 35 лет и старше имеет более высокий риск рождения ребенка с хромосомной аномалией. Это потому, что ошибки в мейозе могут быть более вероятны в результате процесса старения. Женщины рождаются уже со всеми яйцеклетками в яичниках, но они начинают созревать в период полового созревания. Если женщине 35 лет, яйцеклеткам в яичниках также 35 лет.
Вас могут направить на анализ крови на хромосомные патологии при беременности, если вы беременны и старше 35 лет. В мужском организме процессы образования сперматозоидов продолжаются на протяжении всего репродуктивного периода. Поэтому, возраст не сильно увеличивает риск хромосомных нарушений у возрастных отцов.
Но более новые исследования предполагают, что редкие отклонения все же происходят.
Митоз — это деление всех других клеток в организме. Митоз приводит к тому, что количество хромосом в клетке удваивается до 92, а затем клетка делится пополам и количество хромосом также делится пополам — по 46.
Этот процесс постоянно повторяется в клетках по мере роста ребенка. Митоз продолжается на протяжении всей вашей жизни.
Он заменяет клетки кожи, клетки крови и другие типы клеток, которые повреждены или естественным образом погибают.
Во время беременности может возникнуть ошибка в митозе. Если хромосомы не делятся на равные половины, новые клетки могут иметь дополнительную хромосому (всего 47) или недостающую хромосому (всего 45).
Тератоген
Тератоген — это то, что может вызвать или повысить риск врожденного дефекта у ребенка. Это то, с чем может столкнуться мать во время беременности. Тератогены включают в себя:
- Некоторые лекарства
- Наркотики
- Алкоголь
- Табак
- Токсичные химикаты
- Некоторые вирусы и бактерии
- Некоторые виды излучения
Каждая из наших хромосом имеет характерную структуру. Исторически ученые использовали технику окрашивания, которая окрашивает хромосомы в полосатый рисунок. Эти шаблоны полос облегчают идентификацию каждой из наших отдельных хромосом, и благодаря этому можно составить визуальное изображение кариотипа. Любое отклонение от нормального кариотипа известно как аномалия хромосомы.
Половина всех самопроизвольных абортов происходит из-за хромосомных нарушений.
Самые серьезные хромосомные расстройства вызваны потерей или приобретением целых хромосом. Какие это могут быть заболевания:
- Трисомия 21 хромосомы – синдром Дауна (15 на 10 000)
- Трисомия 18 хромосомы – синдром Эдвардса (3 на 10 000)
- Трисомия 13 хромосомы – синдром Патау (2 на 10 000)
- Моносомия Х-хромосомы – синдром Шерешевского-Тернера (2 на 10 000)
- Кариотип XXY – синдром Клайнфельтера (10 из 10 000)
- Кариотип XXX (возможно и большее количество Х-хромосом, например, ХХХХХ) – синдром «суперженщины» (10 на 10000)
- Кариотип XYY (возможно большее количество Y-хромосомы – XYYY) – синдром «супермужчины» (10 на 10000)
- Структурные нарушения могут принимать несколько форм:
- Делеция – утрата части хромосомы, и вследствие этого, части генов, ответственных за те или иные функции в организме
- Дупликация – мутация, вызывающая повторение участка хромосомы, что приводит к дополнительному генетическому материалу.
- Транслокация – мутация, вызывающая перемещение одной части хромосомы в другую часть хромосомы (внутрихромосомно) или в другую хромосому вообще (межхромосомно). Есть два ключевых типа: обратный: сегменты из двух разных хромосом обмениваются частями, и второй — Робертсонский: вся хромосома прикрепляется к другой.
- Инверсия: мутация, какой-то участок хромосомы оказывается повернутым на 180о .
- Кольцевая хромосома – когда в обоих плеча хромосомы отсутствуют конечные фрагменты и они замыкаются, образуя круг
- Изохромосомия – когда в хромосоме фрагменты ДНК повторяются в обоих плечах
Структурные аномалии – это когда большие участки ДНК отсутствуют или лишние, при нормальном количестве хромосом.
Сбалансированные структурные аномалии включают перестройку генетического материала, но без общего прибавления или потери генетического материала, то есть «сумма» неизменна. Например, инверсии и транслокации.
Несбалансированные структурные отклонения связаны с получением или потерей генетического материала – остальные варианты структурных нарушений. Даже крошечные несбалансированные структурные аномалии могут повлиять на многие гены и, следовательно, оказать серьезное влияние на человека.
Чаще всего в практике встречаются транслокации и делеции, нежели другие виды структурных аномалий хромосом.
Несбалансированные структурные отклонения могут быть такими:
- Синдром Вольфа — Хиршхорна – делеция короткого плеча хромосомы 4 (1 на 50 000)
- Синдром кошачьего крика – делеция короткого плеча хромосомы 5 (1 на 50 000)
- Синдром WAGR – микроделеция короткого плеча хромосомы 11 (1 на 500 000 до 1 миллиона)
- Микроделеция Прадера-Вилли / Ангельмана с короткого плеча хромосомы 15 (1 на 15 000)
- Микроделеция Ди-Джорджи из длинного плеча хромосомы 22 (1 в 4000)
Детальная характеристика хромосомной патологии плода при беременности, частой причины врожденных патологий и потери беременности, имеет решающее значение для выяснения генов для развития плода человека.
Среди всех хромосомных аномалий, связанных с самопроизвольным прерыванием беременности, наиболее часто обнаруживаются хромосомные анеуплоидии, которые включают изменение числа копий всей хромосомы, например, трисомию 21, трисомию 13, трисомию 18 и моносомию X, и их встречаемость значительно возрастает с возрастом матери. Хромосомные структурные аномалии, возникающие из-за изменения структуры или частей хромосомы, например, делеции и дупликации, встречаются реже, чем хромосомная анеуплоидия, но скорость их обнаружения значительно улучшилась благодаря применению цитогенетических методов на основе микрочипов.
Рассмотрим немного детальнее некоторые заболевания, которые вызваны нарушениями со стороны количества или качества хромосом.
Пожалуй, самым распространенным заболеванием, о котором знают все, является синдром Дауна. При этом заболевании лишняя 21 хромосома вызывает пороки развития, но, как правило, они совместимы с жизнью, а в реалиях современного уровня медицины серьезные проблемы со здоровьем коррегируются, чем улучшаются прогнозы относительно качества жизни и продолжительности жизни.
Синдром Эдвардс
Синдром Эдвардса – возникает вследствие дополнительной 18 хромосомы. Такие детки рождаются с множественными врожденными пороками развития, причем это чаще девочки. Такие дети маловесные, при том что беременности донашиваются до нормального срока.
Как правило, для синдрома характерны, прежде всего, аномалии строения черепа – он имеет вытянутую форму, и лицевой череп также патологически развивается. Нижняя челюсть недоразвита, может быть расщелина неба и верхней губы, узкие глазные щели, деформированные, низкорасположенные ушные раковины.
Иногда может отсутствовать слуховой проход. Грудная клетка более широкая и короткая, чем у здоровых детей. Характерны и изменения конечностей. Кроме того, аномалии и со стороны внутренних органов – пороки сердца, сосудов, нарушения со стороны формирования мозговых структур, нарушение мышечного тонуса.
К сожалению, несмотря на любое лечение и уход, такие дети умственно не сохранны, интеллект сильно нарушен.
Продолжительность жизни этих детей небольшая – от 3 месяцев до года, но при легких формах заболевания ребенок может прожить несколько лет.
Синдром Патау
Синдром Патау – трисомия 13 хромосомы. Это очень тяжелая хромосомная аномалия, которая приводит к рождению глубоко инвалидизированных детей. Такие дети также рождаются обычно доношенными, но умеренно маловесными.
При беременности характерно многоводие, которое встречается в половине случаев при этой хромосомной патологии.
Из аномалий развития чаще всего встречаются: уменьшение объема мозга – микроцефалия, узкие глаза, возможны аномалии развития глаз в виде циклопии, уменьшении размеров глазных яблок, широкое основание носа, расщелины неба и губы, деформированные ушные раковины, могут быть лишние пальцы на кистях рук и стопах, пороки развития внутренних органов – от порока сердца до удвоения селезенки, почки имеют аномальное строение, половые органы также дефектны. Умственно такие дети не сохранны. Почти все дети с этим синдромом умирают в первый год жизни, но иногда могут жить несколько лет.
Синдром кошачьего крика
Синдром кошачьего крика – патология, которая вызывается делецией короткого плеча 5 хромосомы. Кариотип ребенка при этом — 46 XX или XY, 5р-. При этом выраженность проявлений синдрома зависит не от величины дефекта в хромосоме, а от отсутствия определенного маленького участка хромосомы.
Бывают также случаи мозаицизма, тогда проявления менее выражены. Особенностью детей с таким синдромом, что позволяет предположить патологию еще в родильном доме – характерный «мяукающий» плач ребенка, что происходит из-за аномального строения гортани.
Эта особенность обычно исчезает после первых 12 месяцев жизни. Также характерны маловесность, отставание в развитии, снижение мышечного тонуса, лунообразное лицо и широко расставленные глаза. С синдромом кошачьего крика девочки рождаются немного чаще, чем мальчики.
Кроме того, у таких детей часто бывает микроцефалия, пороки сердца, гипертелоризм, пороки внутренних органов и опорно-двигательной системы. При квалифицированном уходе, медицинском сопровождении прогнозы для жизни достаточно хорошие, но задержка психомоторного и физического развития имеет место.
В любом случае, само по себе заболевание коррекции не поддается, но улучшить качество жизни таких детей вполне возможно.
Cиндром WAGR
Еще один довольно редкий синдром – синдром WAGR. Название этого синдрома – аббревиатура от наиболее частых проявлений заболевания: W – опухоль Вильямса, A – аниридия, G – аномалии со стороны репродуктивных органов, R – отставание в умственном развитии.
Аниридия – это отсутствие радужной оболочки, и обычно сопровождается и другими аномалиями строения и развития органа зрения. Опухоль Вильямса – это нефробластома – очень злокачественная опухоль, которая характерна для детей до 5 лет, пол не важен.
Не всегда при этом генетической патологии наблюдаются сразу все эти признаки, и иногда проявления ограничены не только ними. Иногда этот синдром вообще обнаруживается при обследовании по поводу, например, аниридии.
Изолированное проявление какой-то из составляющих синдрома может и не вызвать подозрения на генетическую патологию, или стать явным уже в более старшем возрасте. Этот синдром не передается по наследству и является следствием спонтанной мутации, поэтому такие дети могут рождаться и у абсолютно здоровых родителей, как впрочем, при почти всех хромосомных аномалиях.
Многие хромосомные патологии могут иметь схожие проявления, и не всегда фенотипически можно сразу точно поставить диагноз. Для этого необходимо провести анализ кариотипа, чтобы иметь возможность давать прогнозы относительно здоровья и жизни таких детей
")




