Ключично-черепной дизостоз: характерные симптомы и лечение дисплазии
» Болезни » Другие » Ключично-черепной дизостоз: характерные симптомы и лечение дисплазии
1807 0
Черепно-ключичный диостоз (дисплазия ключично-черепная, синдром Шейтхауэра-Мари-Сентона) – заболевание, которое относится к смешанным формам заболеваний скелета.
Проблема является наследственной и передаётся по аутосомно-доминантному типу. Заболевание характеризуется полным или частичным отсутствием ключицы, а также проблемами в развитии костей черепа.
Полное отсутствие ключиц в медицинской практике встречается достаточно редко – в 10% случаев. Наиболее часто патология проявляется как отсутствие части ключицы, акромиального отдела и уменьшенных в размерах лопатках.
Причиной развития заболевания есть нарушения, происходящие в одном из генов шестой хромосомы.
Как это выглядит и проявляется
В первую очередь, ключично-черепной дизостоз характеризуется изменениями ключиц, их недоразвитием или полным отсутствием одной.
При данном заболевании человек может сдвинуть ключицы друг к другу таким образом, что впереди они могут друг с другом соприкоснуться.
Изменения происходят и в строении черепа: значительно увеличивается мозговая часть и уменьшена лицевая. Родничок может не закрыться на протяжении всей жизни.
В виду таких внешних характеристик, лицо получается очень маленьким, а лоб и макушка очень большими. Учитывая, что это заболевание генетическое и болеют ним обычно целыми семьями, такие люди чаще всего всем родом занимались цирковой деятельностью.
Верхняя челюсть также претерпевает изменения: твёрдое нёбо укорочено, сама челюсть недоразвита, укорочена в размерах. Развитие постоянных зубов может быть запоздалым: так, молочные зубы могут продержаться до 25-30-летнего возраста.
В результате деформации ключиц, может наблюдаться защемление нервного сплетения, что, в свою очередь, может спровоцировать недоразвитость мышц и общую слабость верхних конечностей.
Но и это ещё не всё, у человека с черепно-ключичной дисплазией может быть:
- задержка роста;
- гипертелоризм – чрезмерно увеличенное расстояние между парными органами (аномально широко посаженные глаза);
- недоразвитие костей таза;
- расщелина неба (или арковидное нёбо);
- позднее прорезывание основных зубов;
- сверхкомпактные зубы, повышенное развитие кариеса;
- уменьшенная грудная клетка;
- кифоз;
- остеосклероз;
- сколиоз;
- в результате пониженной минерализации костей наблюдается их повышенная ломкость;
- в некоторых случаях возможна глухота;
- ассиметричная длина пальцев;
- брахицефалия – короткоголовость;
- умственная отсталость не наблюдается.
Зубной ряд пациента с дизостозом
Сразу после появления младенца на свет о наличии заболевания среди других симптомов может говорить:
- при пальпации кости черепа оказываются мягкими;
- значительно увеличена амплитуда движений (в плечевых суставах) из-за полного или частичного отсутствия ключиц.
К шести-восьмилетнему возрасту для пациентов с данным заболеванием требуется рентгенологическое исследование, так как до этого времени патологий в костно-суставной системе нет.
По достижении шести лет, кости черепа приближаются к норме, в то время как кости таза приобретают более типичный вид для данного заболевания.
Медицинская помощь
Оказание медицинской помощи при данном синдроме включает в себя несколько этапов.
Стоматологическая помощь
Ключично черепной дизостоз – это заболевание, которое не поддаётся лечению, поэтому работа врачей в данном случае заключается в борьбе с проявлениями аномалии.
В детском возрасте наиболее ощутимой есть проблема прорезывания зубов и дальнейшее исправление их положения. По этой причине пациентам с патологией непременно нужно обратиться к ортодонту, который поможет при помощи пластин поставить зубы на свои места.
Далее работа стоматологов заключается в том, чтобы вовремя убрать молочные зубы для более комфортного и скорого прорезывания коренных зубов (если такового не происходит природным путём, врач их обнажает искусственно).
Оперативное вмешательство
Если ключицы отсутствуют полностью, хирургическое вмешательство не назначается из-за нецелесообразности. В таких случаях возможно лишь консервативное лечение.
Если же хотя бы укороченные фрагменты костей присутствуют, оперативный метод имеет место быть. В таком случае будет актуальна операция по восстановлению ключиц.
Оперативное вмешательство – дело сугубо индивидуальное: если пациент (или родители больного) желают устранить проблему, операция будет назначена, если нет, больному предложат консервативное лечение.
ЛФК для лечения и восстановления
Для больных черепно-ключичной дисплазией лечебная физкультура просто необходима, так как ослабленные в результате аномалии верхние конечности необходимо укреплять.
Движения и упражнения должны быть полностью расписаны по индивидуальной программе в зависимости от особенностей течения заболевания и тех участков тела, которые необходимо проработать.
Витаминизация
Приём витаминов для пациентов крайне важен, особенно для тех, кто занимается спортом. Таким образом, происходит укрепление костной ткани и всего организма.
В виду того что иммунитет людей и данной разновидностью дисплазии достаточно низок, его необходимо постоянно поддерживать специальными медицинскими препаратами, а также закаливанием, особенно для профилактики простудных заболеваний.
Профилактика остеопороза
С самого раннего возраста пациент требуется в профилактике остеопороза. В первую очередь необходимо увеличить поступление кальция и витамина D в организм. Обязателен приём солнечных ванн, и приём в пищу печени, сметаны, масла сливочного и морской рыбы.
Профилактика остеопороза — обязательная опция в комплексе медицинской помощи
При отсутствии лечения симптоматика может проявиться более ярко, но вовремя пройденная и повторяющаяся терапия станет залогом нормального функционирования конечностей.
При выборе профессии важно исключить возможность интенсивной нагрузки на плечевой пояс.
Черепно ключичная дисплазия

Многие годы пытаетесь вылечить СУСТАВЫ?
Глава Института лечения суставов: «Вы будете поражены, насколько просто можно вылечить суставы принимая каждый день средство за 147 рублей…
Читать далее »
В первую очередь, ключично-черепной дизостоз характеризуется изменениями ключиц, их недоразвитием или полным отсутствием одной.
НАШИ ЧИТАТЕЛИ РЕКОМЕНДУЮТ!
Для лечения суставов наши читатели успешно используют Sustalaif. Видя, такую популярность этого средства мы решили предложить его и вашему вниманию.
Подробнее здесь…
При данном заболевании человек может сдвинуть ключицы друг к другу таким образом, что впереди они могут друг с другом соприкоснуться.
Изменения происходят и в строении черепа: значительно увеличивается мозговая часть и уменьшена лицевая. Родничок может не закрыться на протяжении всей жизни.
В виду таких внешних характеристик, лицо получается очень маленьким, а лоб и макушка очень большими. Учитывая, что это заболевание генетическое и болеют ним обычно целыми семьями, такие люди чаще всего всем родом занимались цирковой деятельностью.
Верхняя челюсть также претерпевает изменения: твёрдое нёбо укорочено, сама челюсть недоразвита, укорочена в размерах. Развитие постоянных зубов может быть запоздалым: так, молочные зубы могут продержаться до 25-30-летнего возраста.
В результате деформации ключиц, может наблюдаться защемление нервного сплетения, что, в свою очередь, может спровоцировать недоразвитость мышц и общую слабость верхних конечностей.
Но и это ещё не всё, у человека с черепно-ключичной дисплазией может быть:
- задержка роста;
- гипертелоризм – чрезмерно увеличенное расстояние между парными органами (аномально широко посаженные глаза);
- недоразвитие костей таза;
- расщелина неба (или арковидное нёбо);
- позднее прорезывание основных зубов;
- сверхкомпактные зубы, повышенное развитие кариеса;
- уменьшенная грудная клетка;
- кифоз;
- остеосклероз;
- сколиоз;
- в результате пониженной минерализации костей наблюдается их повышенная ломкость;
- в некоторых случаях возможна глухота;
- ассиметричная длина пальцев;
- брахицефалия – короткоголовость;
- умственная отсталость не наблюдается.
Сразу после появления младенца на свет о наличии заболевания среди других симптомов может говорить:
- при пальпации кости черепа оказываются мягкими;
- значительно увеличена амплитуда движений (в плечевых суставах) из-за полного или частичного отсутствия ключиц.
К шести-восьмилетнему возрасту для пациентов с данным заболеванием требуется рентгенологическое исследование, так как до этого времени патологий в костно-суставной системе нет.
По достижении шести лет, кости черепа приближаются к норме, в то время как кости таза приобретают более типичный вид для данного заболевания.
Бронхолегочная дисплазия: что это такое?
Бронхолегочная дисплазия (БЛД) – это хроническое заболевание дыхательной системы у новорожденных, которое возникает при проведении ИВЛ с использованием высоких концентраций кислорода на фоне респираторных нарушений.
Основные проявления – синдром дыхательной недостаточности (ДН) и бронхиальной обструкции, деформация грудной клетки. Основа диагностики бронхолегочной дисплазии – рентгенография ОГК.
Лечение при данной патологии включает в себя неспецифические терапевтические меры: рациональное питание и режим, адекватную респираторную поддержку, симптоматические медикаментозные препараты.
Патогенез
В патогенезе указанных поражений решающее значение отводят незрелости легочной ткани и чрезмерному образованию перекисных соединений на фоне недостаточной активности системы антиоксидантной защиты, что приводит к развитию деструктивных процессов в легких. Дефицит витаминов А и Е хотя и отягощает течение БЛД, но не рассматривается в качестве причины ее развития.
Дополнительным фактором риска служит отек легких в первые дни жизни.
Проведение ИВЛ в интенсивных режимах (при РДС) приводит к развитию «зависимости» от высокой фракционной концентрации кислорода во вдыхаемой смеси (FiO2). При попытке уменьшить максимальное давление на вдохе (PIP) или снижении FiO2 развивается ДН с гипоксемией и гиперкапнией.
Необходимость высокого PIP обусловлена повреждением и деструкцией дыхательных путей (перерастяжение альвеол, нарушение синтеза сурфактанта, повреждение реснитчатого эпителия), уменьшением растяжимости легких из-за фиброза и потери эластических волокон, возникновением интерстициальноой эмфиземы. Зависимость от высокого FiO2 связана с уменьшением количества легочных капилляров и артериол, нарушением газообмена в альвеолах при интерстициальном фиброзе и отеке легочной ткани.
Данные факторы наряду с присоединением инфекции приводят к развитию пневмонии. Пневмония (в сочетании с незрелостью легочной ткани) способствует возникновению интерстициального отека, обструкции нижних отделов дыхательных путей, интерстициального и внутри-альвеолярного фиброза, фокальной эмфиземы, легочной гипертензии.
Черепно-ключичный диостоз (дисплазия, синдром Шейтхауэра-Мари-Сентона): симптомы и лечение

В первую очередь, ключично-черепной дизостоз характеризуется изменениями ключиц, их недоразвитием или полным отсутствием одной.
При данном заболевании человек может сдвинуть ключицы друг к другу таким образом, что впереди они могут друг с другом соприкоснуться.
Изменения происходят и в строении черепа: значительно увеличивается мозговая часть и уменьшена лицевая. Родничок может не закрыться на протяжении всей жизни.
В виду таких внешних характеристик, лицо получается очень маленьким, а лоб и макушка очень большими. Учитывая, что это заболевание генетическое и болеют ним обычно целыми семьями, такие люди чаще всего всем родом занимались цирковой деятельностью.
Верхняя челюсть также претерпевает изменения: твёрдое нёбо укорочено, сама челюсть недоразвита, укорочена в размерах. Развитие постоянных зубов может быть запоздалым: так, молочные зубы могут продержаться до 25-30-летнего возраста.
В результате деформации ключиц, может наблюдаться защемление нервного сплетения, что, в свою очередь, может спровоцировать недоразвитость мышц и общую слабость верхних конечностей.
Но и это ещё не всё, у человека с черепно-ключичной дисплазией может быть:
- задержка роста;
- гипертелоризм – чрезмерно увеличенное расстояние между парными органами (аномально широко посаженные глаза);
- недоразвитие костей таза;
- расщелина неба (или арковидное нёбо);
- позднее прорезывание основных зубов;
- сверхкомпактные зубы, повышенное развитие кариеса;
- уменьшенная грудная клетка;
- кифоз;
- остеосклероз;
- сколиоз;
- в результате пониженной минерализации костей наблюдается их повышенная ломкость;
- в некоторых случаях возможна глухота;
- ассиметричная длина пальцев;
- брахицефалия – короткоголовость;
- умственная отсталость не наблюдается.
Зубной ряд пациента с дизостозом
Сразу после появления младенца на свет о наличии заболевания среди других симптомов может говорить:
- при пальпации кости черепа оказываются мягкими;
- значительно увеличена амплитуда движений (в плечевых суставах) из-за полного или частичного отсутствия ключиц.
К шести-восьмилетнему возрасту для пациентов с данным заболеванием требуется рентгенологическое исследование, так как до этого времени патологий в костно-суставной системе нет.
По достижении шести лет, кости черепа приближаются к норме, в то время как кости таза приобретают более типичный вид для данного заболевания.
Синдром Тричера-Коллинза – лечение
Не существует медикаментозного метода, позволяющего вылечить челюстно-лицевой дизостоз: операция является единственным методом терапии. Хирургическое лечение назначают как с эстетической целью, так и для нормализации работы челюсти, чтобы пациент смог принимать пищу. При необходимости осуществляется коррекция ушных раковин, пластика наружного слухового прохода.
Из-за маленького размера челюстей и большого языка он просто не может поместиться во рту. В таком случае удаляют надгортанник и устанавливают постоянную трахеостому. Пациенту нередко требуется проведение нескольких операций из-за сложности патологии. Как выглядит синдром Тричера-Коллинза до и после операции изображено на фото.
Диагностика заболевания
На рентгенограмме чаще всего хорошо просматриваются охваченные преждевременным синостозированием лямбдовидный, сагиттальный и коронарный швы.
Помимо этого могут наблюдаться такие рентгенологические находки как маленькие параназальные синусы, расширение ямки гипофиза, базилярный кифоз, уплощение глазниц и пальцевые вдавления.
Томографическое исследование четко указывает на деформацию внутреннего слухового прохода.
Томограмма височной кости обнаруживает наружную ротацию каменистого отдела пирамиды, вторичную относительно дисплазии основания черепа. Вследствие этого имели косое направление слуховые каналы, направление лицевого нерва была неправильным и наблюдались гиперостозы. Причем к первичным изменениям следует относить аномалии слуховых косточек, закрытие овального окна.
Томография височной кости обнаруживает отсутствие барабанной полости, атрезию или стеноз наружного слухового прохода, сужение и искривление воздухоносных пространств сосцевидного отростка и среднего уха, деформацию стремени, анкилоз молоточка с внешней стенкой верхнего отдела барабанной полости. Помимо этого прослеживается недоразвитие периостальной части лабиринта.
Дифференцированная диагностика проводится с такими заболеваниями как синдром Сетре-Чотцена, синдром Пфейффера и синдром Апера.
Симптомы
Такой необычный недуг имеет много самых разных проявлений. Причём у одного человека с этим диагнозом могут наблюдаться далеко не все возможные дефекты. А так как это заболевание врождённое, то первые признаки болезни можно наблюдать сразу после рождения ребёнка.
Ключично черепная дисплазия — диагноз, как проявляется, лекарства, расшифровка

Диагностика ключично-черепного дизостоза основана на клинических симптомах и рентгенологических исследованиях, которые включают изображения черепа, грудной клетки, таза и рук.
Главный рентгенологический симптомом — дефекты ключиц. Обычно отсутствует наружный (акромиальный) конец ключицы, в то время как внутренний (грудинный) конец присутвует. Но иногда ключица состоит из двух фрагментов.
Полное отсутствие ключицы встречается редко.
При молекулярно-генетическом исследовании обнаруживаются мутации гена RUNX2 у 60 % −70 % людей с диагнозом ключично-черепной дизостоз.
Первый случай отклонений в развитии ключиц был описан М. Мартин в 1765 году. Случай, связанный с отклонениями в развитии ключиц и костей черепа был рассмотрен в 1861 году Шейтхауэром.
В 1897 году Мари и Сентон более подробно рассмотрели это заболевание, указали на семейный характер патологии, и дали название dyostosis cleido-cranialis, поскольку соотносили его с дефектами костей черепа и ключиц.
Гессе в 1926 году описал отклонения в развитии зубов и челюстей и связал его с ключично-черепным дизостозом.
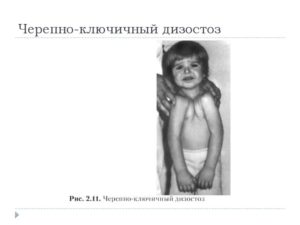
Ключично-черепной дизостоз: характерные симптомы и лечение дисплазии
Рис. 1. Ключично-черепной Дизостоз (синдром Шейтхауэра—Мари—Сентона) у трех сестер. Отсутствие ключиц у двух сестер приводит к полному соприкосновению плеч.
Ключично-черепной Дизостоз (синдром Шейтхауэра — Мари — Сентона) характеризуется гипоплазией покровных костей черепа в сочетании с полным или частичным недоразвитием одной или обеих ключиц, т. е. нарушением развития так наз. мембранозных костей. Для Д. этого вида характерно незаращение или позднее заращение черепных швов и родничков, брахицефалия (см.
) с преобладанием расширения свода черепа в латеральных направлениях, выдающийся лоб, гипоплазия лицевых костей, гл. обр. верхней челюсти, обусловливающая псевдопрогению (кажущееся увеличение нижней челюсти).
Нарушение развития челюстей сопровождается запаздыванием прорезывания зубов.
Отсутствие ключиц или частичное недоразвитие их с дефектом внутренних, средних или наружных частей ведет к увеличению подвижности плечевого пояса, а при полном отсутствии их — к полному соприкосновению плеч (рис. 1).
Описанные изменения часто сопровождаются деформациями позвоночника, костей верхних и нижних конечностей, стоп, тазовых костей. Аномалия наследуется по рецессивному и доминантному типу, может быть семейной.
При ключично-черепном дизостозе рентгенологически выявляются многочисленные изменения со стороны скелета, однако наиболее характерны изменения ключиц и костей черепа.
Дефекты ключиц чаще симметричны и могут быть разных размеров: от небольших до полного отсутствия ключиц. Чаще же всего отсутствует акромиальный конец ключицы.
Свободный конец оставшейся части закруглен, покрыт замыкающей костной пластинкой и связан плотным фиброзным тяжем с акромиальным отростком лопатки. По ходу фиброзного тяжа иногда обнаруживаются костные включения.
При рентгенол, исследовании черепа определяется брахицефалия: мозговой череп увеличен в поперечнике и уменьшен в передне-заднем размере. Основание черепа укорочено в поперечном направлении и несколько удлинено в продольном. Кости свода, особенно лобная, истончены и как бы раздуты, значительно выдаваясь в стороны. Передний родничок остается незаращенным.
При исследовании скелета туловища и конечностей могут быть обнаружены отклонения в развитии ряда костей: уменьшенные размеры лопаток, крестца, костей таза с отсутствием слияния между собой лобковых, седалищных и подвздошных костей и недоразвитием лобкового симфиза; недоразвитие проксимальных отделов бедер с варусной деформацией их; укорочение или отсутствие ногтевых бугристостей у концевых фаланг пальцев кистей и стоп; незаращение дужек позвонков.
При множественном поражении скелета наличие характерных изменений ключиц делает рентгенол, диагноз достоверным.
Симптоматика
Основные симтомы ключично-черепного дизостоза:
- Недоразвитие или отсутствие одной или обеих ключиц. При отсутствии или недоразвитии ключицы плечевой пояс резко сужен, надплечья покаты и опущены. Отмечается избыточная подвижность в плечевых суставах, возможно даже соприкоснуться плечами спереди грудины.
- Задержка закрытия (окостенения) пространства между костями черепа (родничков), могут формироваться дополнительные костные включения. Большой родничок может оставаться открытым в течение всей жизни.
- Нарушения формирования корней, задержка в прорезывании молочных и постоянных зубов. Могут до 25-30-летнего возраста не меняться молочные зубы. Часто встречаются сверхкомплектные зубы.
Также в большинстве случаев отмечено:
- Низкий рост по сравнению с родственниками.
- Брахицефалия.
- Гипертелоризм.
- Высокий и выдающийся вперед лоб.
- Недоразвитие костей таза.
Другие медицинские проблемы включают рецидивы инфекций верхних дыхательных путей; осложнения, рецидивы инфекции ушей; ранний остеопороз и проблемы с суставами; высокую частоту кесарева сечения у женщин; легкую степень моторной задержки у детей в возрасте до пяти лет.
У больных классическим ключично-черепным дизостозом нормальный уровень интеллекта.
Заболевание встречается поровну у мужчин и женщин, и ему подвержены все расы.
Челюстно-лицевой Дизостоз
Челюстно-лицевой Дизостоз (синдром Берри—Франческетти, синдром Франческетти—Цвалена) — гипоплазия гл. обр. нижней челюсти и скуловых костей, макростомия (своеобразное «рыбье» или «птичье» лицо), широкие косо расположенные глазные щели (рис.
3), с вывороченными и скошенными книзу веками и колобомами в наружных отделах, слепые фистулы от углов рта к ушам, языковидное оволосение щек, нарушения развития зубов, деформация ушных раковин, иногда среднего и внутреннего уха с развитием глухоты, устранимой операцией.
Челюстно-черепной Дизостоз
Челюстно-черепной Дизостоз (синдром Петерс — Хевельса) — гипоплазия верхней челюсти, скуловых дуг, открытый прикус, прогения (выстояние нижней челюсти), укорочение переднего отдела основания черепа. Аномалия наследуется по доминантному типу.
Существуют другие формы черепных Д.: синдромы Гегенхара, Робена, Франсуа и др. Внешний вид больных с различными формами Д. характерен. Д. сохраняется всю жизнь, не поддается оперативной коррекции, почти не требует дифференциальной диагностики с другими заболеваниями. В сомнительных случаях важным диагностическим методом является рентгенол, исследование.
Различают так наз. неполные типы перечисленных Д., когда имеют место не все характеризующие их симптомы. Отдельные признаки могут комбинироваться в различных сочетаниях, составляя как бы промежуточные типы Д.
Прогноз для жизни благоприятный.
Библиография: Алексеев В. А. Случай черепно-ключичного дизостоза, Вестн, рентгенол, и радиол., № 3, с. 80, 1974; Косинская Н. С. Нарушения развития костно-суставного аппарата, с. Зв и др., Л., 1966; КручинскийГ. В. Редкие врожденные синдромы лица и челюстей (в границах первой и второй жаберных дуг), Минск, 1974, библиогр.; P e й н-б e р г С. А.
Рентгенодиагностика заболеваний костей и суставов, кн. 1—2, М., 1964; Ромоданов А. П. и JI я гц e н-к о Д. С. Черепно-лицевой Дизостоз, Журн, невропат, и психиат., т. 72, № 10, с. 1487, 1972, библиогр.; Fleischer-Peters A. Kiefermissbildungen bei Dysostose-Syndromen des Schadels, Dtsch, zahnarztl. Z., Bd 24, S. 932, 1969; Humange-netik, hrsg. v. P. E.
T. П. Виноградова; И. Г. Лагунова (рент.).
Черепно-лицевой Дизостоз
Рис. 2. Однояйцовые близнецы 13 лет с черепно-лицевым дизостозом (синдром Крузона). Характерны широко расставленные глаза, выражено косоглазие, гипоплазия верхней челюсти.
Черепно-лицевой Дизостоз (синдром Крузона, гипертелоризм) — недоразвитие костей черепа, мозга и верхней челюсти в сочетании с преждевременным закрытием черепных швов, экзофтальмом (см.), косоглазием (см.), нистагмом (см.), расстройством зрения.
Лоб в области переносицы бугрист, глаза широко расставлены (рис. 2), ное своеобразной крючковидной формы («клюв попугая»), гипоплазия верхней челюсти, псевдопрогения; в резко выраженных случаях наблюдается снижение умственного развития.
Наследуется по доминантному типу.
Рентгенологически выявляются изменения черепа.
На первый план выступает характерная деконфигурация головы и нарушение нормальных соотношений между мозговым и лицевым черепом: первый уменьшен в размерах, имеет почти шаровидную форму, швы заращены, усилены пальцевые вдавления.
Кости свода черепа истончены, несколько выпячиваются кнаружи в области переднего родничка. Основание черепа укорочено и углублено, область турецкого седла сужена, глазницы уплощены.
Кости лицевого черепа малы: верхняя челюсть и носовые кости недоразвиты, нижняя челюсть значительно выдается вперед, в силу чего образуется резкий прогиб носа внутрь.
Рис. 3. Ребенок с челюстно-лицевым дизостозом. Характерны широкие косо расположенные глазные щели, нарушение развития зубов.
САМОЕ ИНТЕРЕСНОЕ:
Люди с Синдромом Тричера Коллинза (3 фото 3 видео)
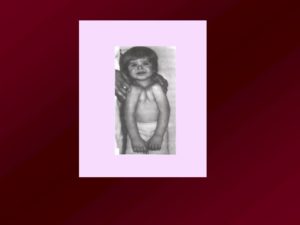
Синдром Тричера – генетическое заболевание, на возникновение которого в большинстве случаев не влияют никакие внешние или внутренние факторы.
Можно сказать, что патология изначально заложена в аминокислотный код будущего ребенка и начинает проявляться задолго до его рождения. Научно доказано, что спонтанные изменения в структуре ДНК (генные мутации) у лиц, имеющих синдром, возникают в 5 хромосоме.
Последняя является самой длинной нуклеотидной структурой в геноме человека и отвечает за производство материала для скелета плода.
Происходят мутации по причине сбоя внутриклеточного синтеза белка. В результате чего развивается синдром гаплонедостаточности. Последний характеризуется нехваткой белка, необходимого для правильного развития лицевой части черепа. При всем этом следует знать, что болезнь Тричера-Коллинза имеет аутосомно-доминантный, реже – аутосомно-рецессивный характер.
- этанол и его производные;
- цитомегаловирус;
- радиоактивное излучение;
- токсоплазмоз;
- прием противосудорожных и психотропных препаратов, лекарств с ретиноевой кислотой.
Диагностика
Диагностика ключично-черепного дизостоза основана на клинических симптомах и рентгенологических исследованиях, которые включают изображения черепа, грудной клетки, таза и рук.
Главный рентгенологический симптомом — дефекты ключиц. Обычно отсутствует наружный (акромиальный) конец ключицы, в то время как внутренний (грудинный) конец присутвует. Но иногда ключица состоит из двух фрагментов.
Полное отсутствие ключицы встречается редко.
При молекулярно-генетическом исследовании обнаруживаются мутации гена RUNX2 у 60 % −70 % людей с диагнозом ключично-черепной дизостоз.
Пренатальное исследование челюстно-лицевых аномалий проводится на 10-11 неделе беременности при помощи биопсии ворсин хориона. Процедура в достаточной степени опасная, поэтому в дородовой диагностике синдрома Тричера врачи предпочитают использовать УЗИ. Кроме того, у членов семьи берутся анализы крови.
Постнатальная диагностика проводится на основании имеющихся клинических проявлений. При полной экспрессивности синдрома Тричера вопросов, как правило, не возникает, чего нельзя сказать, когда обнаруживаются незначительные признаки данной патологии. В этом случае проводится комплексная диагностика состояния, включающая следующие исследования:
- оценку и мониторинг эффективности кормления;
- аудиологическое тестирование слуха;
- рентгеноскопию черепно-лицевой дисморфологии;
- пантомографию;
- КТ или МРТ головного мозга.
Аналогичные методы исследования применяются, когда необходимо провести дифференциальную диагностику для того, чтобы распознать неярко выраженные проявления болезни Тричера-Коллинза и отличить их от признаков других патологических состояний. Так, в большинстве случаев специалисты назначают дополнительные инструментальные исследования для дифференциации указанного недуга с синдромами Гольденхара (гемифациальной микросомии), Нагера.
Какие мутации приводят к развитию синдрома Тричера Коллинза
Чаще всего при синдроме Тричера Коллинза происходит мутация в генах TCOF1, POLR1C и POLR1D. При этом изменения в гене TCOF1 обнаруживается в 93% всех случаев постановки этого диагноза.
Мутация в генах POLR1C и POLR1D выявляется довольно редко. Именно это является причиной развития синдрома Тричера Коллинза.
Если же нарушений в этих генах нет, но заболевание присутствует, то причину его можно считать неизвестной.
Известно, что эти три гена — TCOF1, POLR1C, и POLR1D – играют важную роль в формировании костей и других тканей лицевой части черепа. Они принимают активное участие в производстве молекул, которые носят название рибосомная РНК, которая является «сестрой» ДНК.
Изменения в вышеперечисленных генах сокращают общее количество производимых молекул. Предполагают, что это приводит к самоуничтожению некоторых клеток, которые отвечают за развитие тканей лица и черепа. Всё это ещё во время формирования плода приводит к тому, что в формировании лица есть некоторые проблемы, которые могут быть как едва заметными, так и сильно выраженными.
Болезнь Тричера-Коллинза имеет три стадии. На начальном этапе его развития наблюдается незначительная гипоплазия лицевых костей. Вторая стадия характеризуется деформацией и недоразвитостью слуховых проходов, маленькой нижней челюстью, аномалиями глазной щели, что прослеживается практически на всех фото пациентов с синдромом.
История
Первый случай отклонений в развитии ключиц был описан М. Мартин в 1765 году. Случай, связанный с отклонениями в развитии ключиц и костей черепа был рассмотрен в 1861 году Шейтхауэром.
В 1897 году Мари и Сентон более подробно рассмотрели это заболевание, указали на семейный характер патологии, и дали название dyostosis cleido-cranialis, поскольку соотносили его с дефектами костей черепа и ключиц.
Гессе в 1926 году описал отклонения в развитии зубов и челюстей и связал его с ключично-черепным дизостозом.
Черепно-ключичный дизостоз: причины появления и лечение заболевания. Ключично-черепной дизостоз: характерные симптомы и лечение дисплазии
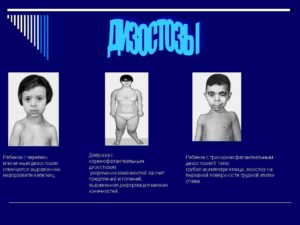
Основными симптомами наличия данной патологии являются:
- отсутствие или недоразвитие обеих или одной из ключиц, из-за чего плечевой пояс сильно сужен, а надплечья опущены. По этой же причине в плечевых суставах заметна чрезмерная подвижность;
- задержка окостенения родничков, при этом в некоторых случаях формируются дополнительные костные включения. В некоторых случаях большой родничок так и не закрывается на протяжении всей жизни человека.;
- присутствие нарушений в формировании корней зубов, а также задержки при прорезывании постоянных и молочных зубов. Иногда у людей с этой патологией молочные зубы не меняются до 30 лет, но при этом отмечается наличие сверхкомплектных зубов.
В большинстве случаев отмечаются также такие симптомы, как брахицефалия, низкий рост (в сравнении с родственниками), гипертелоризм, недоразвитие костей таза, выдающийся вперед и высокий лоб.
У пациентов с классической формой этой патологии обычно нормальный уровень интеллекта, но существует ряд других медицинских проблем. Так, такие люди подвержены рецидивам и осложнениям инфекций ушей, дыхательных путей, у них рано появляются проблемы с суставами и развивается остеопороз, а у детей отмечают легкую степень моторной задержки.
Челюстно-лицевой дизостоз – симптомы
Выявить челюстно-лицевой дизостоз можно уже при первом осмотре пациента:
- У таких детей отмечается выраженная недоразвитость скуловых костей, челюсти.
- Ушные раковины полностью не сформированы: уши у ребенка имеют малый размер, а слуховой канал недоразвит.
- Разрез глаз заметно сужается.
- Тип и выраженность нарушения может варьироваться, однако всегда происходит сбой в работе слухового аппарата, дыхательной системы.
Самым ярким симптом, сопровождающим болезнь Тричера-Коллинза, является изменение нормальной формы глаз. Глазная щель сужается, в результате чего глазные яблоки опускаются. Возможны и другие проявления синдрома Тричера-Коллинза:
- дефект мягкой ткани ротовой полости;
- впалый подбородок;
- нарушение слуха;
- нарушение прикуса;
- расщепление верхнего неба;
- изменение прикуса.
Синдром Тричера-Коллинза – степени
Как отмечалось выше, патология развивается постепенно, прогрессируя со временем. Синдром Тричера-Коллинза в начальной степени характеризуется незначительной гипоплазией лицевых костей. Диагностируется уменьшение в размерах скуловых костей, из-за чего лицо выглядит слегка вытянутым. Такое изменение именуют первой степенью заболевания.
Челюстно-лицевой дизостоз или синдром Тричера-Коллинза второй степени сопровождается недоразвитием слуховых проходов. Нижняя челюсть значительно меньшего размера, глазная щель сужается, что приводит к нарушению зрения. При тяжелой форме лицо практически отсутствует. Нос, скуловые кости, верхняя и нижняя челюсти деформируются настолько, что пациент становится неузнаваемым для окружающих.
Медицинский центр Бейт Гейди
ПозвонитьНаписать
Все клиники
Синдром Тричера-Коллинза – прогноз
Синдром Тричера-Коллинза является тяжелым испытанием для пациентов. Прогноз полностью зависит от степени выраженности деформации лицевых костей, количества сопутствующих клинических нарушений. В большинстве случаев патологии прогноз благоприятный. Но дети с данным заболеванием нередко сталкиваются с трудностями социальной адаптации, что негативно отражается на общем самочувствии.
")
")



