Синдром Прадера-Вилли
Синдром Прадера-Вилли – это редкое генетическое заболевание, характеризующееся грубыми конституциональными нарушениями, когнитивными и психическими расстройствами. Клиническая картина разнообразна, основные симптомы включают ожирение, задержку роста и умственную отсталость.
Часто встречается снижение мышечного тонуса, репродуктивная дисфункция. Окончательный диагноз устанавливается на основании молекулярно-генетического исследования. Специфическое лечение не разработано.
Осуществляется симптоматическая терапия по основным компонентам синдрома: назначение гипокалорийной диеты и гормональных средств, индивидуальные занятия с дефектологом и т. д.
Синдром Прадера-Вилли (синдром гипотонии-ожирения) является одной из наиболее выраженных форм генетически обусловленного ожирения. Заболевание впервые было описано в 1956 году швейцарскими педиатрами А. Прадером и Х. Вилли.
Несмотря на генетическую природу, болезнь носит спорадический характер. По разным статистическим данным, распространенность синдрома составляет 1:15 000 – 1:25 000 новорожденных. Какие-либо значимые гендерные различия отсутствуют.
Синдром Прадера-Вилли
Патология развивается в результате мутации 15 хромосомы (сегмента q11.2-q13). Прямой передачи заболевания по наследству не происходит. Хромосомная аномалия возникает в момент оплодотворения яйцеклетки, т. е.
обмена родительских генетических материалов. В 65-75% случаев мутация обусловлена дефектом отцовской 15 хромосомы, а в 25-35% – наследованием обеих 15 хромосом от матери.
Факторы риска, провоцирующие клинические проявления хромосомной мутации, неизвестны.
Патологические механизмы остаются малоисследованными. Известно, что при этой болезни наблюдается выраженный дисбаланс между процессами липолиза и синтеза жиров в подкожно-жировой клетчатке со сдвигом в сторону последнего. Предполагается, что ведущую роль в ожирении и задержке роста у детей с синдромом Прадера-Вилли играет эндокринная дисрегуляция.
Дисфункция ядер гипоталамуса приводит к снижению выработки многих гормонов, таких как соматотропный гормон, гонадотропины, тиреотропный гормон и пр. Падение концентрации гормона роста и половых гормонов, особенно в детском возрасте, способствует накоплению жировых депо. Характерно повышение уровня пептидного гормона грелина, который является эндогенным стимулятором аппетита.
В генезе нейропсихических расстройств рассматривается роль низкого уровня нейротрофического фактора головного мозга, участвующего в развитии и дифференцировке клеток центральной нервной системы и их функциональной активности. Гипопигментация кожи и волос объясняется подавленной функцией тирозиназы в волосяных фолликулах и меланоцитах.
Клинические проявления начинают манифестировать уже в период внутриутробного развития. Отмечается малая подвижность плода, неправильное предлежание, недоношенность при рождении.
Возникает выраженная мышечная гипотония. Значительно ослаблены сосательный и глотательный рефлексы. Это затрудняет кормление ребенка и ведет к недостаточному возрастному набору массы тела.
В некоторых случаях необходимо питание через зонд.
Несколько позже присоединяется наиболее характерный симптом – полифагия (патологически повышенный аппетит), вследствие которой ребенок довольно быстро начинает прибавлять в весе, достигая ожирения, вплоть до морбидного. Отложение жира преимущественно происходит в области туловища и проксимальных отделах конечностей.
Выражены нейропсихические нарушения. Речь замедлена, интеллектуальные способности (память, концентрация внимания, последовательная обработка информации) значительно отстают от возрастной нормы.
В подростковом периоде нередко наблюдаются обсессивно-компульсивные расстройства, резкие перепады настроения, агрессивное поведение.
Из-за недостаточной продукции слюны зубы быстро поражаются кариесом.
Гипогонадизм у мальчиков проявляется гипоплазией мошонки, микропенисом, крипторхизмом, у девочек – недоразвитием половых губ, поздним наступлением менструаций или их полным отсутствием.
Возможны нарушения координации, мышечные судороги, косоглазие. Из других конституциональных изменений можно отметить низкий рост, акромикрию (уменьшенный размер кистей и стоп).
Типичны гипопигментация кожи, светлые волосы.
Преобладающее число осложнений синдрома Прадера-Вилли связано с морбидным ожирением. Избыток жировой массы способствует раннему развитию инсулинорезистентности, метаболического синдрома и сахарного диабета 2 типа. Нередко встречается неалкогольная жировая болезнь печени (жировой гепатоз). Значительное скопление жира в области шеи обуславливает сужение просвета дыхательных путей.
Вследствие этого более чем у половины пациентов (55-60%) наблюдается синдром обструктивного апноэ сна, который в свою очередь, резко увеличивает риск артериальной гипертензии, инсульта, жизнеугрожающих аритмий. Ожирение также вызывает альвеолярную гиповентиляцию и чрезмерную нагрузку на правые отделы сердца, в результате чего возникает правожелудочковая сердечная недостаточность.
Из-за сниженной минеральной плотности костной ткани любая травма может привести к переломам. Практически все больные страдают первичным бесплодием. Отмечаются частые вирусные инфекции верхних дыхательных путей, бронхиты и пневмонии. Существуют данные о том, что при синдроме ПВ повышается вероятность развития лейкемии и других онкологических заболеваний.
Больных, страдающих синдромом Прадера-Вилли, курируют врачи-педиатры и генетики. При общем осмотре обращают внимание на ослабление мышечного тонуса и сухожильных рефлексов, конституциональные изменения – ожирение, низкий рост. Дополнительное обследование включает следующие исследования:
- Анализы крови. В биохимическом анализе нередко обнаруживается повышение концентрации глюкозы и печеночных трансаминаз (АЛТ, АСТ). Отмечается снижение уровня гонадотропинов (ФСГ, ЛГ), половых гормонов (тестостерона, эстрогенов), соматотропного гормона.
- Денситометрия. При проведении двойной энергетической рентгеновской абсорбциометрии определяются признаки остеопении или остеопороза – показатели плотности костей ниже среднего значения пиковой костной массы более чем на 2,5 SD.
- Определение наличия СОАС. Поскольку обструктивное апноэ представляет угрозу для здоровья и жизни, все пациенты с подозрением на синдром Прадера-Вилли проходят кардиореспираторный мониторинг и полисомнографическое исследование, при которых обнаруживаются высокий индекс дыхательных расстройств и индекс десатурации.
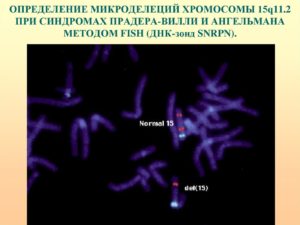
- Генетическое исследование. Выявление микроделеции 15q11-13 с помощью полимеразной цепной реакции, кариотипирования или флуоресцентной гибридизации – основной верифицирующий тест, позволяющий достоверно поставить диагноз.
Дифференциальный диагноз проводится с заболеваниями, которые сопровождаются выраженной мышечной гипотонией и задержкой нейропсихического развития – синдромом Опица-Фриаса, миопатиями, спинальной амиотрофией. Кроме того, синдром ПВ дифференцируется с другими наследственно обусловленными формами ожирения (адипозогенитальная дистрофия, синдром Лоуренса-Муна).
Пациенты подлежат госпитализации в педиатрическое отделение. Эффективные методы этиотропной терапии не разработаны, все лечебные мероприятия носят симптоматический характер. Для борьбы с гипотонией назначаются сеансы массажа и физиотерапевтические методы воздействия. Рекомендуются занятия с логопедом, дефектологом, психотерапевтом. Другие виды лечения синдрома Прадера-Вилли:
- Диета. Основное внимание уделяется изменениям в питании. Необходимо ограничить продукты с высоким содержанием насыщенных жиров и легкоусвояемых углеводов. Общий суточный калораж должен составлять 1000-1200 ккал. Лекарственные препараты, подавляющие аппетит, не используются, так как показали низкую эффективность у больных синдромом ПВ.
- Заместительная гормональная терапия. Рекомендуется подкожное введение рекомбинантного соматотропного гормона даже в раннем детском возрасте еще до наступления ожирения. Для восстановления репродуктивной функции применяются аналоги гонадотропин-рилизинг гормона (гозерелин).
- СИПАП-терапия. Для лечения синдрома обструктивного апноэ наиболее успешным методом является использование специального устройства для автоматической интраназальной вентиляции легких, создающего постоянное положительное давление в верхних дыхательных путях.
- Антиостеопоротическое лечение. При низких показателях плотности костей во избежание патологических переломов назначаются витамин Д (холекальциферол), препараты кальция, бисфосфонаты (золедроновая кислота).
Хирургическое лечение
При наличии определенных показаний (удлиненное мягкое небо, гипертрофия миндалин) для устранения СОАС выполняется хирургическая коррекция – увулопалатофарингопластика, которая заключается в иссечении части мягкого неба, тонзиллэктомии, формировании швов, подтягивающих заднюю стенку глотки. Вероятность рецидива после операции составляет около 50%.
Если не удается добиться снижения массы тела консервативными методами, прибегают к бариатрической хирургии – бандажированию желудка, желудочному шунтированию. Сохранение крипторхизма к концу 1-го года жизни служит показанием к оперативному устранению патологии. Проводится орхипексия – прикрепление яичка к мошонке с помощью швов.
Экспериментальное лечение
Ведутся разработки новых лекарственных средств для терапии синдрома ПВ. Имеются обнадеживающие результаты клинических исследований применения агониста рецепторов окситоцина – карбетоцина.
Предлагается воздействовать на кишечную микробиоту больных детей пробиотическими препаратами.
В экспериментальных работах на лабораторных животных продемонстрировало лечебный эффект вещество UNC0642, активирующее гены на необходимом участке 15 хромосомы.
Продолжительность жизни пациентов, страдающих синдромом ПВ, при своевременной диагностике и адекватном лечении достигает 60-70 лет. В отсутствие превентивных мер смерть может наступить в возрасте 4-5 лет от сердечно-легочной недостаточности. В 50% случаев причиной летального исхода становится обструктивное апноэ сна и вызванные им сердечно-сосудистые катастрофы.
Реже больные погибают от тяжелой респираторной инфекции. Единственным способом предотвращения возникновения заболевания является пренатальная диагностика и прерывание беременности. Основная роль отводится вторичной профилактике – предупреждению осложнений болезни, например вакцинации от гриппа и пневмококковой инфекции.
Синдром Прадера-Вилли: выраженные симптомы, выбор лечения, прогноз

Синдром Прадера-Вилли — редкое наследственное заболевание, причиной которого является отсутствие отцовской копии участка хромосомы 15q11-13.
В этом участке хромосомы 15 находятся гены, в регуляции которых задействован геномный импринтинг.
Большинство случаев является спорадическими, для редких описанных семейных случаев характерно неменделевское наследование. Частота встречаемости — 1 : 12 000-15 000 живорождённых младенцев.
Общие сведения
Впервые синдром Прадера-Вилли упоминается Лэнгдоном Дауном в 1887 г. при описании 14-летней пациентки, страдающей задержкой роста, ожирением, снижением умственной деятельности и функции яичников (гипогонадизмом). Лэнгдом Даун назвал это заболевание полисарцией.
Классическое описание синдрома появилось в 1956 г. благодаря исследованиям пациентов с подобным фенотипом, проведенным швейцарскими педиатрами Андреа Прадером, Гвидо Фанкони, Генрихом Вилли и терапевтом Алексисом Лабхартом.
В 1965 г. был выявлен синдром Ангельмана, при котором мутации в этом же генетическом регионе (15q11-13) затрагивают не отцовскую, а материнскую копию хромосомы.
В 1981 г. при изучении 15-й хромосомы Ледбеттер и соавторы определили регион хромосомы, который при мутациях является причиной синдрома Прадера – Вилли.
Частота распространения заболевания колеблется от 1 к 10 000 до 1 к 25 000 новорожденных.
Заболевание одинаково часто наблюдается у мальчиков и девочек, и является основной генетически обусловленной причиной ожирения у лиц старше 1 года. Национальность и раса на распространенность заболевания не влияют.
Генетический механизм На сегодняшний день точно установлено, что при данной патологии 15-я хромосома повреждается в сегментах от q11.2 до q13. То же самое происходит при синдроме Ангельмана. Однако данное заболевание характеризуется совершенно иными симптомами.
Подобный диссонанс могут объяснить лишь только таким явлением в генетической науке, как геномный импринтинг, а также унипарентальная дисомия. При унипарентальной дисомии обе хромосомы наследуются лишь от одного родителя, но для того чтобы это произошло, на генный материал должны влиять определенные биохимические факторы.
Этот факт установлен при помощи прометафазных анализов и ДНК-маркирования некоторых локусов данной хромосомы. Синдром Прадера-Вилли обусловлен двумя основными механизмами: микроделецией 15-й хромосомы, полученной от отца, и идиосомией материнских хромосом, обе из которых получены от матери.
При геномном импринтинге изменения фенотипа зависят от того, в чьих хромосомах – отца или матери — произошла экспрессия.
Симптоматика
Симптомы у синдрома Прадера-Вилли различные, меняются в зависимости от возрастной категории больного.
Внутриутробно патологическое состояние имеет такие признаки:
- пониженная активность движений плода;
- необычное расположение;
- многоводие.
Сразу после рождения симптоматика следующая:
- ягодичное предлежание плода;
- пониженное давление;
- снижение сосательного рефлекса;
- затрудненное дыхание.
В детстве у ребенка отмечают такие состояния:
- заторможенное развитие навыка речи;
- поражение зубов кариесом;
- угнетенная координация движений;
- употребление большого количества пищи;
- быстрый набор лишнего веса;
- проблемы со сном;
- сколиоз;
- половое созревание наступает позже обычного;
- невысокий рост;
- задержка интеллектуального развития;
- задержка психомоторного развития;
- чрезмерная гибкость.
Признаки в совершеннолетнем возрасте:
- бесплодие;
- небольшое количество волос в области лобка;
- ожирение;
- склонность к сахарному диабету.
Обобщенная симптоматика внешних отличий у взрослых людей включает:
- нос больших размеров, широкий;
- узкие пальцы на руках;
- конечности маленького размера;
- присутствует гиперчувствительность кожного покрова;
- большое количество избыточного веса;
- лоб высокий и узкий;
- кожный покров и волосы более светлого оттенка по сравнению с родственниками.
Наблюдается задержка моторного и полового развития.
Диагностические мероприятия
Ранняя диагностика синдрома Прадера-Вилли и последующее лечение позволяют улучшить прогноз развития заболевания. Диагноз ставится, как правило, на основе клинических проявлений болезни, но на сегодняшний день часто используется генетическое тестирование, которое специалисты рекомендуют в первую очередь для новорожденных.
Это обусловлено тем фактом, что у детей наличие синдрома определить намного труднее, поскольку невозможно проверить их способности, позволяющие проводить диагностику синдрома Прадера-Вилли по клиническим проявлениям. Генетическое тестирование проводится методом ДНК-метилирования с целью выяснения, присутствуют ли на 15 хромосоме отклонения, приводящие к возникновению заболевания.
Данный способ диагностики синдрома Прадера-Вилли помогает выявить 97% случаев болезни. Стоит также отметить, что часто заболевание диагностируется неправильно, поскольку его нередко путают с синдромом Дауна, который встречается гораздо чаще. К тому же такой характерный признак синдрома Прадера-Вилли, как ожирение, может присутствовать также при синдроме Дауна.
По этой причине огромное число случаев болезни остаются не выявленными.
Лечение
Поскольку синдром Прадера — Вилли относится к врожденным генетическим аномалиям, специфические способы его лечения отсутствуют.
Терапия направлена на нормализацию обмена жиров и углеводов, а также на устранение симптомов заболевания.
При лечении больных используются:
- диетотерапия, включающая учет калорийности пищи и контроль за режимом питания;
- оральные гипогликемические препараты для лечения сахарного диабета;
- кломифен, восстанавливающий секрецию гонадотропинов и половых стероидов;
- гормон роста.
На раннем этапе заболевания при гипотонусе назначается массаж.
При выявлении нарушений биоэнергетического обмена назначаются витамины (Е, С, В2, тиамин, витамин РР), коэнзим Q10, улучшающий кровообращение циннаризин.
Медикаментозное лечение дополняется занятиями с логопедом и дефектологом.
Дети с синдромом Прадера – Вилли постоянно находятся под наблюдением эндокринолога, невролога, психотерапевта и офтальмолога.
Профилактика синдрома
Предотвратить врожденное заболевание невозможно, главное в этом случае – не допустить появления осложнений. Лечение синдрома следует начать как можно раньше, тогда ребенку будет проще приспособиться к обучению в школе и к жизни в обществе.
К профилактике заболевания можно отнести медико-генетические консультации семей, у которых есть предрасположенность к возникновению синдрома. Будущим родителям необходимо провести дородовое генетическое исследование, которое поможет определить особенности строения хромосом плода.
Чтобы улучшить жизнь ребенка с СПВ, следует обеспечить постоянное сотрудничество специалистов медицинских учреждений, родителей и самого малыша.
Прогноз
Среди людей, у которых имеется синдром, значительно повышены показатели соматической заболеваемости, затруднено общение, возникает потребность в специфической помощи, обусловленной особенностями их заболевания. Они могут не понимать, зачем необходимо заботиться о своем здоровье.
Если состояние удовлетворительно и больной чувствует себя хорошо, качество жизни его улучшается. Необходимо устранить следующие факторы: Повышенный риск внезапной смерти. Вероятность заболеваения. Увеличение количества факторов, которые определяют материальное благополучие. Недостаточный доступ к оздоровительным услугам и медицинскому обслуживанию.
Люди с патологией Прадера-Вилли имеют особые потребности, обусловленные их основным заболеванием. Они нуждаются в особой терапии острых и хронических патологий, в помощи в укреплении общего здоровья и т. д.
Их потребности должны быть удовлетворяться в специальных учреждениях, обеспечивающих медицинскую помощь, которая, в свою очередь, может состоять в лечении основного заболевания и соматических нарушений, связанных с основной патологией.
Какова продолжительность жизни с синдромом Прадера-Вилли? Данное заболевание часто приводит к снижению длительности жизни больных до 60 лет. Однако прогноз на выздоровление таких людей весьма неутешительный. В статье было представлено подробное описание синдрома Прадера-Вилли. Теперь вы знаете, что это за патология.
(1 5,00 из 5)
Загрузка…
")
")



