Синдром Клайнфельтера: мужчина с женской хромосомой

Елена Шведкина об одном из самых распространенных генетических заболеваний — больные жалуются на бесплодие, эректильную дисфункцию, гинекомастию и остеопороз
Синдром Клайнфельтера — генетическое заболевание, характеризующееся дополнительной женской половой хромосомой Х (одной или даже несколькими) в мужском кариотипе ХY. При этом в мужских половых железах — яичках — образуется недостаточно половых гормонов.
Как известно, генетический набор человека насчитывает 46 хромосом, из которых 22 пары называются соматическими, а 23‑я пара — половая.
Женщины имеют пару половых хромосом ХХ, а мужчины — ХY.
Для синдрома Клайнфельтера обязательно наличие мужской Y-хромосомы, поэтому, несмотря на дополнительные Х-хромосомы, пациенты всегда являются мужчинами.
Классификация: виды кариотипов при синдроме Клайнфельтера
По количеству дополнительных Х-хромосом различают следующие варианты синдрома Клайнфельтера:
- 47,ХХY — наиболее часто встречающийся
- 48,ХХХY
- 49,ХХХХY
Кроме того, к синдрому Клайнфельтера также относят мужские кариотипы, включающие, помимо дополнительных Х-хромосом, дополнительную Y-хромосому — 48,ХХYY. И, наконец, среди пациентов с этим синдромом встречаются лица с мозаичным кариотипом 46,ХY/47,ХХY (то есть часть клеток имеет нормальный хромосомный набор).
История открытия синдрома
Синдром получил свое название в честь Гарри Клайнфельтера — врача, в 1942 году впервые описавшего клиническую картину болезни. Клайнфельтер с коллегами опубликовали отчет об обследовании 9 мужчин, объединенных общими симптомами, такими как слабое оволосение тела, евнухоидный тип телосложения, высокий рост и уменьшенные в размерах яички.
Позднее, в 1956 г., генетики Планкетт и Барр (Е. R. Plankett, М. L. Barr) обнаружили у мужчин с синдромом Клайнфельтера тельца полового хроматина в ядрах клеток слизистой оболочки полости рта, а в 1959 году Полани и Форд (P. E. Polanyi, S. E.
Ford) с сотрудниками показали, что у больных в хромосомном наборе имеется лишняя Х-хромосома.
Активные исследования данной патологии велись в 70‑х годах в США. Тогда всех новорожденных мальчиков подвергали кариотипированию, в результате чего удалось достоверно выявить распространенность и генетические особенности синдрома Клайнфельтера.
Любопытно, что мыши также могут иметь синдром трисомии по половым хромосомам XXY, что позволяет эффективно использовать их в качестве моделей для исследования синдрома Клайнфельтера.
Распространенность заболевания
Синдром Клайнфельтера является одним из наиболее распространенных генетических заболеваний: на каждые 500 новорождённых мальчиков приходится 1 ребёнок с данной патологией.
Кроме того, синдром Клайнфельтера — третья по распространенности эндокринная патология у мужчин (после сахарного диабета и патологии щитовидной железы) и наиболее частая причина врожденного нарушения репродуктивной функции у мужчин.
На сегодняшний день около половины случаев синдрома Клайнфельтера остаются нераспознанными. Часто такие пациенты обращаются за помощью по поводу бесплодия, эректильной дисфункции, гинекомастии, остеопороза, анемии и пр. без установленного ранее диагноза.
Этиология и причины нарушения
Синдром Клайнфельтера относится к генетическим заболеваниям, не передающимся по наследству, поскольку больные, за редким исключением, бесплодны.
Патология, как правило, возникает в результате нарушения расхождения хромосом на ранних стадиях формирования яйцеклеток и сперматозоидов. При этом синдром Клайнфельтера, возникающий за счет нарушения в женских половых клетках, встречается в три раза чаще.
Мозаичные формы обусловлены патологией деления клеток на ранних стадиях эмбриогенеза, поэтому часть клеток у таких пациентов имеет нормальный кариотип.
Причины нерасхождения половых хромосом и нарушения деления клеток на самых ранних стадиях эмбриогенеза до сих пор малоизучены. В отличие от других хромосомных заболеваний, влияние возраста родителей отсутствует или выражено незначительно.
Ранние признаки
В отличие от большинства заболеваний, связанных с нарушением количества хромосом, внутриутробное развитие детей с синдромом Клайнфельтера проходит нормально, склонности к преждевременному прерыванию беременности не наблюдается.
Так что в младенческом и раннем детском возрасте заподозрить патологию практически невозможно. Более того, клинические признаки классического синдрома Клайнфельтера проявляются, как правило, только в подростковом периоде.
Однако есть симптомы, которые позволяют заподозрить наличие синдрома Клайнфельтера в препубертатном периоде:
- высокий рост (пик прибавки роста приходится на период между 5–8 годами);
- длинные ноги (непропорциональное телосложение);
- высокая талия.
У части пациентов наблюдается некоторая задержка в развитии речи.
В подростковом возрасте синдром часто проявляется гинекомастией, которая при данной патологии имеет вид двустороннего симметричного безболезненного увеличения грудных желез. Так как такого рода гинекомастия часто наблюдается у совершенно здоровых подростков, этот симптом часто остается без внимания.
В норме подростковая гинекомастия бесследно исчезает в течение нескольких лет, у пациентов же с синдромом Клайнфельтера обратной инволюции грудных желез не происходит.
В некоторых случаях гинекомастия может не развиваться вовсе, и тогда патология проявляется признаками андрогенной недостаточности уже в постпубертатный период.
Симптомы андрогенной недостаточности при синдроме Клайнфельтера
Андрогенная недостаточность при синдроме Клайнфельтера связана с постепенной атрофией яичек, что приводит к снижению синтеза тестостерона. Степень недостаточности андрогенов резко варьирует.
В первую очередь обращают на себя внимание внешние признаки гипогонадизма:
- скудная растительность на лице или же полное ее отсутствие;
- рост волос на лобке по женскому типу;
- волосы на груди и других частях тела отсутствуют;
- маленький объем яичек (2–4 мл) и их плотная консистенция (патогномоничный признак).
Поскольку дегенерация половых желез, как правило, развивается в постпубертатный период, у большинства пациентов размеры мужских половых органов, за исключением яичек, соответствуют возрастным нормам.
Пациенты могут жаловаться на ослабление либидо и снижение потенции.
У многих мужчин с синдромом Клайнфельтера половое влечение вовсе не возникает, а некоторые — напротив, заводят семью и живут нормальной половой жизнью.
Наиболее постоянный признак патологии — бесплодие, именно оно чаще всего становится причиной обращения таких пациентов к врачу. У 10 % мужчин с азооспемией обнаруживают синдром Клайнфельтера.
Всем пациентам с нарушениями сперматогенеза необходимо определять кариотип для исключения или подтверждения диагноза синдрома Клайнфельтера.
Недостаток андрогенов приводит к развитию остеопороза, анемии и слабости скелетной мускулатуры. У трети больных можно наблюдать варикозное расширение вен голеней.
Андрогены влияют на обмен веществ, поэтому больные с синдромом Клайнфельтера склонны к ожирению, нарушению толерантности к глюкозе и сахарному диабету второго типа.
Доказана предрасположенность таких пациентов к аутоиммунным заболеваниям (ревматоидный артрит, системная красная волчанка, аутоиммунные заболевания щитовидной железы и другие).
Психологические особенности
Коэффициент интеллекта у больных с классическим синдромом Клайнфельтера варьирует от значений ниже среднего до показателей, значительно превышающих средний уровень.
Однако во всех случаях отмечается диспропорция между общим уровнем интеллекта и вербальными способностями, так что нередко пациенты с достаточно высоким IQ испытывают трудности при восприятии больших объемов материала на слух, а также при построении фраз, содержащих сложные грамматические конструкции.
Такие особенности причиняют пациентам много неприятностей в период обучения и нередко продолжают сказываться на профессиональной деятельности.
Данные о психологических особенностях больных с синдромом Клайнфельтера достаточно противоречивы, однако большинство специалистов оценивают пациентов как скромных, робких людей с несколько заниженной самооценкой и повышенной чувствительностью.
Есть данные, свидетельствующие о склонности пациентов с синдромом Клайнфельтера к гомосексуализму, алкоголизму и наркомании.
Сложно сказать, вызваны ли особенности психики у таких больных непосредственным влиянием хромосомной аномалии, или же это реакция на проблемы в сексуальной сфере.
В отношении разных цитогенетических вариантов синдрома Клайнфельтера справедливо правило, что с увеличением количества дополнительных Х-хромосом увеличивается количество и выраженность патологических симптомов.
Диагностика синдрома Клайнфельтера
Во многих странах синдром Клайнфельтера часто диагностируется ещё до рождения ребёнка, так как многие женщины позднего детородного возраста, в связи с высоким риском генетических дефектов у будущего потомства, используют пренатальную генетическую диагностику плода. Нередко пренатальное выявление синдрома Клайнфельтера является поводом для прерывания беременности, в том числе и по рекомендации врачей. В России анализ кариотипа будущего ребёнка проводится крайне редко.
При подозрении на синдром Клайнфельтера проводят лабораторный анализ крови для определения уровня мужских половых гормонов. Необходима дифференциальная диагностика с другими заболеваниями, протекающими с проявлениями андрогенной недостаточности. Точный диагноз синдрома Клайнфельтера ставят на основании изучения кариотипа (набора хромосом) больного.
Исследования, необходимые для подтверждения диагноза
| Анализы | Результаты |
| Кариотип | 47,ХХY (80 % случаев) 48,ХХYY 48,ХХХY 49,ХХХХY 46,ХY/47,ХХY |
| Концентрация ЛГ, ФСГ | Повышена, особенно ФСГ |
| Концентрация общего тестостерона | Чаще снижена (в некоторых случаях нормальная за счет повышения секс-стероид-связывающего глобулина СССГ или на начальной стадии развития заболевания) |
У всех мужчин с резко повышенными концентрациями гонадотропинов необходимо исключить синдром Клайнфельтера, так как нередко первый лабораторный признак этой генетической патологии — повышение в крови концентрации гонадотропинов при нормальном содержании общего тестостерона.
Синдром Клайнфельтера необходимо дифференцировать от других форм первичного гипогонадизма. В любом случае при повышении уровня ФСГ в крови необходимо определение кариотипа для исключения в первую очередь синдрома Клайнфельтера.
Цели лечения синдрома Клайнфельтера:
- Восстановление нормального содержания тестостерона
- Восстановление сексуальной функции
- Ликвидация метаболических нарушений
При клинически выраженной патологии необходима пожизненная заместительная терапия препаратами тестостерона.
Адекватная терапия позволяет не только улучшить внешний вид и общее самочувствие больного, но и вернуть способность к нормальной половой жизни. Кроме того, заместительная терапия предупреждает развитие остеопороза, купирует мышечную слабость. В юном возрасте лечение необходимо начинать сразу же после постановки диагноза.
При синдроме Клайнфельтера лучше использовать препараты тестостерона длительного действия:
- смесь эфиров тестостерона в виде масляного раствора, инъекции которого необходимо делать 2–3 раза в месяц;
- тестостерона ундеканоат в виде масляного раствора — препарат-депо с замедленным высвобождением действующего вещества — инъекции 1 раз в 3 месяца.
Гормонолечение при наличии Х хромосомы у мужчин должно носить постоянный характер. Дозу препарата подбирают индивидуально под контролем уровня тестостерона и ЛГ в сыворотке крови.
Уже развившаяся гинекомастия при синдроме Клайнфельтера не подвергается инволюции даже в случае адекватного лечения, поэтому часто приходится прибегать к хирургической коррекции (мастэктомии).
Для профилактики таких сопутствующих заболеваний, как ожирение и сахарный диабет второго типа, больным рекомендуют придерживаться диеты и следить за собственным весом.
Мониторинг пациентов с синдромом Клайнфельтера следует осуществлять не реже 1 раза в 6–12 месяцев. Он должен включать следующие исследования:
- общий анализ крови для оценки уровня гемоглобина и гематокрита;
- гормональный анализ крови, включающий определение тестостерона и ЛГ (проводится на фоне лекарственной терапии за 1–2 дня до очередной инъекции тестостерона);
- денситометрию (всем пациентам, у которых на момент постановки диагноза были обнаружены остеопения или остеопороз).
Внедрение интрацитоплазматической инъекции сперматозоида в яйцеклетку (ИКСИ) и данные о возможности присутствия зародышевых клеток в яичках у пациентов с синдромом Клайнфельтера предопределили применение метода искусственного оплодотворения для данной категории пациентов, некоторые попытки были удачными.
Прогноз
Прогноз для жизни и трудовой деятельности у пациентов с классическим синдромом Клайнфельтера — в целом благоприятен. Ранняя заместительная терапия, психологическая работа с пациентами и их родителями позволяют больным полностью адаптироваться в современном обществе.
Синдром Алажилля: клинические рекомендации, симптомы

Психотерапевт высшей категории Олег Викторович
47197
Дата обновления: Март 2020
К числу редких генетических патологий относится синдром Алажилля. Заболевание напрямую связано с аномалиями со стороны гепатобилиарной системы.
У людей с таким диагнозом наблюдается недостаточное количество желчных протоков, которые предназначены для выведения из печени выработанной желчи. Болезнь имеет тяжелое течение.
На благоприятный прогноз ее лечения могут рассчитывать пациенты, у которых диагностирован синдром в легкой форме.
Что такое синдром Алажилля?
Мутация гена JAG1 вызывает развитие пороков печени, костной ткани и сердца и является главной причиной артериопеченочной дисплазии (синдрома Алажилля)
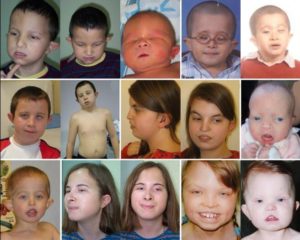
Синдромом Алажилля называют синдромальную патологию, которая включает в себя не меньше 3 из 5 главных признаков, характерных для данного заболевания. Речь идет о дефектах глаз и сердечно-сосудистой системы, врожденной форме гипоплазии и черепно-лицевых аномалиях.
Болезнь указана в международной классификации болезней МКБ-10 под кодом Q44.7. Он отведен для других врожденных аномалий печени.
Заболевание встречается 1 раз на 70 000 случаев. С патологией сталкиваются живорожденные дети в разных странах мира.
Специалисты выделяют легкую и тяжелую степень синдрома Алажилля. Они отличаются между собой выраженностью клинических признаков и вариантом лечения.
Причины нарушения
Артериопеченочная дисплазия (еще одно название синдрома Алажилля) относится к группе генетических заболеваний. Патология передается по аутосомно-доминантному пути. Это значит, что у человека с таким диагнозом будут болеть синдромом все дети.
причина развития болезни – мутация гена JAG1. Именно она вызывает развитие пороков печени, костной ткани и сердца.
Симптоматика
У человека с синдромом Алажилля диагностируется помутнение роговицы глаза
Заболевание имеет специфические проявления, которые позволяют врачам диагностировать его у пациентов. Симптомы синдрома Алажилля могут варьироваться в пределах семьи.
| Характерные симптомы | |
| Печень | Практически все признаки поражения имеют отношение к плохому оттоку желчи. Их можно наблюдать с первых дней жизни человека. У пациентов развивается желтуха, темнеет моча и повышается уровень билирубина в крови. |
| Кожный покров и мягкие ткани | Из-за того, что в крови человека накапливается слишком большое количество билирубина, у него появляется сильный зуд. Данный симптом возникает примерно к третьему месяцу жизни ребенка с генетическим отклонением. Под кожными покровами у пациентов нередко образуются скопления жировых отложений. Их в медицине называют ксантомами. К появлению данных скоплений приводит повышенный уровень в крови холестерина. Новообразования напоминают своим внешним видом небольшие узелки разной формы. Как правило, они окрашены в желтый цвет. Чаще всего ксантомы образуются в области локтей, на животе и коленях. Данные уплотнения не несут в себе прямой угрозы для жизни человека. |
| Рост и обмен веществ | У людей с данным заболеванием часто случается диарея. Это состояние связано с неспособностью усвоения нутриентов органами желудочно-кишечного тракта. Это состояние также может негативно отражаться на росте у детей. Больные с генетическим отклонением нередко сталкиваются с переломами костей. У них наблюдаются проблемы со зрением и памятью. Не исключается явная задержка полового созревания. |
| Сердце | Врачи диагностируют у пациентов с таким диагнозом аномальное сужение артерий легких, которые доставляют кровь к этому органу от сердца. У определенного процента больных данная патология способна привести к порокам развития внутреннего органа. |
| Лицо | Выдают заболевание глубоко посаженые глаза. Этот симптом встречается практически у 96% пациентов. Также у них можно заметить выступающий лоб и подбородок, оттопыренные уши и прямой нос. Характерные для патологии черты не сильно бросаются в глаза только в первые несколько лет жизни ребенка. |
| Глаза | У человека с синдромом Алажилля диагностируется помутнение роговицы глаза. Данная аномалия не имеет никакого влияния на остроту зрения. |
| Костная система | Не исключается аномальная форма костей позвоночника. Эту патологию удается обнаружить в процессе проведения рентгенографии и во время изучения готовых снимков. Крайне редко нарушение приводит к развитию проблем со спиной. |
| Почки | При генетическом заболевании присутствует множество патологий почек. У больных с таким диагнозом удается диагностировать стеноз артерий, гипоплазию и образование кист во внутреннем органе. Чаще всего присутствует почечная недостаточность. |
| Селезенка | Из-за проблем в работе печени может нарушиться кровоток. В результате этого страдает селезенка, которая выполняет функцию очищения крови и ее передачи непосредственно в фильтрующий орган. Подобные проблемы приводят к развитию портальной гипертензии и увеличению размера селезенки. |
| Кровеносные сосуды | Нередко у больных синдромом Алажилля присутствуют аномалии со стороны сонных артерий. По этой причине они попадают в группу риска развития инсульта и внутренних кровотечений. Не исключается возникновение аномалий и в других артериях. |
С течением болезни ее клиническая картина значительно усугубляется.
Опасен ли синдром Алажилля?
У маленьких детей с синдромом Алажилля быстро развивается цирроз печени
Заболевание может привести к развитию осложненных состояний, если не будет начато его своевременное лечение. В данном случае прогноз выздоровления для пациента окажется неблагоприятным.
Осложнения проявляются у пациентов, у которых наблюдается отсутствие большого количества желчных ходов, из-за чего внутренний орган не может обеспечить нормальный отток желчи.
В течение 1-2 лет после рождения у ребенка активно развивается цирроз печени. Данное заболевание приводит к летальному исходу, так как оно вызывает полное нарушение работы печени.
Передается ли нарушение по наследству?
Патология характеризуется аутосомно-доминантным типом наследования. Это значит, что она передается по наследству от больного родителя к ребенку. Генный дефект напрямую связан с делецией 20-й хромосомы.
Аномальное изменение ранее получалось верифицировать в 3,6% случаев при помощи современных методов исследования. Благодаря новейшим разработкам в области молекулярно-генетических исследований верификацию удалось увеличить до 70%.
Диагностика
При подозрении на развитие синдрома Алажилля требуется провести полную диагностику больного. Она включает в себя следующие этапы исследования:
- Сбор анамнеза. Специалист определит аномальное увеличение размера живота, которое присутствует с рождения ребенка. Также будут отмечены симптомы желтухи и кожного зуда.
- Получение данных осмотра пациента. Врач выявит сочетание врожденных пороков разных внутренних органов и костных структур. Обращается внимание на характерную внешность больного и его отставание в развитии.
- Изучение наследственности. В ходе диагностики выявляется присутствие аномалии у одного из родителей ребенка.
- Биопсия тканей печени. Это обязательное исследование, которое помогает определить наличие признаков отсутствия внутрипеченочных желчных ходов. В процессе диагностики специалист делает забор кусочка внутреннего органа и проводит его анализ в лабораторных условиях.
- Проведение биохимического анализа крови. Исследование предоставляет информацию о повышении количества билирубина, липидов и холестерина в организме больного.
- Генетическое исследование. Анализ выявляет наличие мутаций в гене JAG1, которые характерны для синдрома Алажилля.
В ходе диагностики может потребоваться консультация генетика и окулиста.
В обязательном порядке проводится дифференциальная диагностика, которая помогает отличить синдром от других патологических процессов со стороны гепатобилиарной системы.
Возможно ли лечение синдрома Алажилля?
Капсулы принимают вечером, не разжевывая, запивая небольшим количеством воды, пациентам с массой тела менее 34 кг рекомендуется прием препарата в виде суспензии
Традиционные методы лечения, которые позволяют дать бой генетическим болезням, направлены на коррекцию осложнений, вызванных длительным холестазом. Народные методы для лечения синдрома крайне нежелательно использовать, так как они могут усугубить текущее положение пациента.
Для людей с синдромом Алажилля подбирается специальное питание, которое отличается высоким содержанием среднецепочечных триглицеридов.
В борьбе с кожным зудом, от которого страдают дети старшей возрастной группы, могут использоваться соответствующие препараты. Их применяют строго по назначению врача.
Пациентам показана трансплантация печени, если у них диагностированы патологические состояния, которые отрицательно сказываются на жизни человека. В таком лечении нуждаются больные с жалобами на постоянный зуд, физическое отставание в развитии и дефицит в жирорастворимых витаминах.
В обязательном порядке проводится медикаментозная терапия. Она предусматривает применение урсодезоксихолевой кислоты, микроэлементов и жирорастворимых витаминов.
Субтеломерные делеции 1q43q44 и тяжелые поражения головного мозга, связанные с задержкой миелинизации | журнал генетики человека
- перегласовка
- Миелин биология и ремонт
- Нарушения развития нервной системы
Субтеломерные делеции 1q44 вызывают задержку умственного развития, задержку развития и аномалии головного мозга, в том числе аномалии мозолистого тела (ACC) и микроцефалию у большинства пациентов.
Мы сообщаем о случаях шести пациентов с делециями 1q44; два пациента с интерстициальными делециями 1q44; и четыре пациента с терминальными делециями 1q. У одного из пациентов наблюдалась несбалансированная транслокация между хромосомой 5.
Все области делеции перекрывались с ранее сообщенными критическими областями для ACC, микроцефалии и судорог, что указывает на рецидивную природу основных фенотипических признаков делеций 1q44.
У четырех пациентов с терминальными делециями 1q обнаружена сильная потеря объема в мозге по сравнению с пациентами, у которых были интерстициальные делеции 1q44. Это указывало на то, что теломерные области играют важную роль в серьезной потере объема мозга.
Кроме того, у двух пациентов с терминальными делециями 1q43, за пределами критической области для синдрома делеции 1q44, наблюдалась отсроченная миелинизация. Поскольку области делеции, идентифицированные у этих пациентов, расширяются к центромере, мы заключаем, что гены, ответственные за задержанную миелинизацию, могут быть расположены в соседней области 1q43.
Субмикроскопические субтеломерные хромосомные делеции были обнаружены у 7, 4% детей с умственной отсталостью от умеренной до тяжелой степени. 1 Некоторые субтеломерные делеционные синдромы являются клинически узнаваемыми и идентифицируются по характерным признакам, тогда как некоторые другие не могут быть идентифицированы с помощью таких средств.
Недавнее развитие молекулярного кариотипирования с использованием хромосомных микрочипов выявило четкие корреляции генотип-фенотип и выявило критические хромосомные области с характерными признаками субтеломерных делеций. Наиболее ярким примером является синдром Миллера-Дикера, который показал четкие корреляции генотип-фенотип.
2 Синдром Миллера-Дикера вызывается субтеломерной делецией 17p и хорошо распознается и характеризуется лиссэнцефалией и отличительными чертами лица 3, которые являются результатом участия гена регуляторной субъединицы 1 фактора активации тромбоцитов 1b ( PAFAH1B1 ) и тирозина Ген 3-монооксигеназы / триптофана 5-монооксигеназы-активирующий эпсилон-полипептидный ген ( YWHAE ) соответственно; оба эти гена расположены на 17p13. 3
Несколько исследований исследовали критическую область для синдрома субтеломерной делеции 1q44 и обнаружили, что основными фенотипическими признаками синдрома субтеломерной делеции 1q44 являются микроцефалия, аномалии мозолистого тела (ACC) и судороги. 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 Недавно Ballif et al. 17 проанализировали пациентов с микроделециями 1q44 и предложили определенные гены, которые могут отвечать за индивидуальные особенности.
Мы сообщаем о случаях шести вновь выявленных пациентов с делециями 1q44; два с интерстициальной делецией 1q44; и четыре с удалением терминала 1q. Поскольку у пациентов с терминальной делецией 1q44 обнаружены более тяжелые фенотипы по сравнению с пациентами с интерстициальной делецией, фенотипические различия будут получены из дополнительно удаленной области 1q43q44.
Хромосомные делеции
Тестирование хромосомных микрочипов выявило аберрации в области 1q43q44 у шести пациентов (рис. 1). У пациентов 1 и 2 были удалены интерстициальные делеции 1, 9 и 2, 2 Мб, соответственно, в области 1q43q44. У пациента 1 анализ FISH подтвердил удаление (рис. 2а).
Последующий анализ FISH не выявил аномалий у родителей обеих семей, что указывает на делеции de novo у обоих пациентов.
Молекулярное кариотипирование определяло аберрации как arr 1q44 (243 809 193–245 665 521) × 1 дн для пациента 1 и arr 1q43q44 (243 303 991–245 506 920) × 1 дн для пациента 2.
Результаты тестирования хромосомных микрочипов, представленные в Gene View Agilent Genomic Workbench (Agilent Technologies). Вертикальная ось и горизонтальная ось представляют отношение сигналов log 2 и геномное положение соответственно. Аберрантные области показаны синими прямоугольниками. Точки указывают положения генома и отношение log 2 каждого зонда.
Изображение в полном размере
Результаты анализов FISH. ( a ) Потеря зеленой маркировки сигнала RP11-7L23 (стрелка) указывает на удаление 1q44 у пациента 1. ( b ) Потеря зеленой маркировки сигнала RP11-88N11 (стрелка) указывает на удаление 1q44 у пациента 3.
( c — e ) Подтверждение несбалансированной транслокации между хромосомой 1 и 5 у пациента 4. Потеря зеленой маркировки сигнала RP11-143E8 (стрелка) указывает на делецию 1q44 ( c ). Дополнительная сигнальная метка RP11-94J21 5p15.
33 присутствует на другой хромосоме ( d, красный сигнал, стрелка), что указывает на транслокацию на хромосому 1 ( e, зеленый сигнал, стрелка).
Изображение в полном размере
У пациента 3 терминальные делеции 1q43 были идентифицированы и подтверждены с помощью анализа FISH (рис. 2b). Поскольку анализ FISH для обоих родителей не выявил никаких отклонений, эта делеция произошла как de novo . Молекулярное кариотипирование пациента 3 было обозначено как обр 1q43q44 (242 442 098–249 250 621) × 1 дн.
У пациента 4 была выявлена потеря числа копий генома в 1q43q44 и дополнительное усиление в 5p15.33 (дополнительная фигура 1). Последующий анализ FISH подтвердил несбалансированную транслокацию между 1q43 и 5p у пациента 4 (Рисунки 2c-e), и ни у одного из родителей транслокация не была обнаружена.
Следовательно, было установлено, что транслокация дисбаланса у пациента была de novo по происхождению. Ее кариотип был 46, XX, дер (1) т (1; 5) (q43; с15, 33) .arr 1q43q44 (242 223 230–249 212 668) × 1, 5p15, 33 (57 640–1 705 515) × 3 дн.
Дублированная область 5p была только 1, 7 Мб из терминальной области.
У пациента 5 терминальные делеции 1q43 были идентифицированы с точкой разрыва в гене гомолога 3 вирусного онкогена v-akt мышиной тимомы ( AKT3 ). Молекулярный кариотип был arr 1q44 (243 880 099–249 212 668) × 1. Наибольшая делеция была выявлена у пациента 6 с arr 1q43q44 (238 888 870–249 212 668) × 1.
Результаты анализа FISH приведены в дополнительной таблице 1.
Клинический отчет
9-летний мальчик родился вакуумной экстракцией. Его масса тела при рождении составляла 2820 г (в пределах 25-го центиля), длина составляла 45 см (= 3-й центиль), а окружность затылочно-лобного отдела тела (OFC) составляла 32, 5 см (10-25-й центиль).
После рождения у него появились проблемы с кормлением. Его развитие было слегка задержано: контроль над головой был достигнут через 8 месяцев, сидя без поддержки в 15 месяцев, ползая через 24 месяца и ходя в одиночестве без поддержки через 44 месяца.
В возрасте 11 месяцев он перенес рецидив фебрильных судорог. В 18 месяцев он перенес нефебрильные судороги.
Магнитно-резонансная томография головного мозга (МРТ) в возрасте 4 лет показала потерю объема в лобной доле, легкие аномальные формы гираля в лобной доле и боковой доле, а также ACC (Рисунки 3a – c). Обычный хромосомный анализ выявил нормальный мужской кариотип.
Результаты МРТ головного мозга пациента 1, осмотренного в 4 года ( a — c ), пациента 2, осмотренного в 3 года ( d — f ), пациента 3, осмотренного в 12 месяцев ( g — i ), пациента 4, осмотренного в 15 месяцев ( j — 1 ) пациент 5 обследован через 5 месяцев ( м- о ), а пациент 6 обследован через 3 года ( р- р ). T1-взвешенные сагиттальные виды ( a, d, g, j, m, p ), T1-взвешенные осевые виды ( b, e, h, k, n, q ) и T2-взвешенные осевые виды ( c, f, i, l, o, r ). АКК отмечены у всех пациентов. Пациент 5 ( м ), в частности, демонстрирует полный агенез мозолистого тела. Уменьшение объема лобной доли наблюдается у всех пациентов. Заметно отсроченная миелинизация отмечена у пациентов 3 и 4 ( i и l ).
Изображение в полном размере
В настоящее время его рост составляет 119, 5 см (
")




