Миопатия — симптомы, лечение, причины болезни, первые признаки
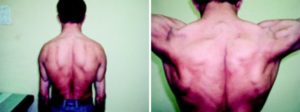
Миопатия – заболевание, которое возникает вследствие нарушения в метаболизме и строении мышечной ткани, в результате чего появляется снижение силы пораженных мышц, а также происходит ограничение двигательной активности.
Миопатии относятся к группе нервно-мышечных заболеваний, для которой характерно дистрофическое поражение мышечной ткани с атрофией отдельных волокон (миофибрилл) в некоторых местах.
Кроме этого, происходит замещение атрофированных миофибрилл соединительной или жировой тканью.
Это приводит к утрате мышцами способности к сокращению, что обуславливает появление в клинике заболевания мышечной слабости, а также ограничение двигательной активности.
Выделяют две основные формы заболевания:
- Первичные миопатии, которые зачастую имеют наследственный характер. В свою очередь они делятся на следующие виды:
- врожденные миопатии, которые развиваются в младенческом возрасте;
- ранние детские миопатии, которые развиваются в возрасте 5-10 лет;
- юношеские миопатии, которые развиваются в подростковом возрасте.
- Вторичные миопатии, возниающие на фоне других заболеваний.
Например, на фоне эндокринных расстройств (болезнь Иценко-Кушинга, гиперальдостеронизм, гиперпаратиреоз, гипо- и гипертиреоз), хронических интоксикаций (алкоголизм, наркомания, работа на предприятиях с профессиональными вредностями), тяжелых хронических заболеваний (хроническая почечная недостаточность, хроническая печеночная недостаточность, хроническая обструктивная болезнь легких, сердечная недостаточность), а также опухолевых процессов.
Первичные формы миопатии встречаются значительно чаще, чем вторичные. Поэтому крайне важно во время планирования беременности проконсультироваться у генетика для того, чтобы выявить риск развития данной патологии у будущего ребенка.
Исходя из того, в какой части тела мышечная слабость выражена больше, выделяют следующие виды миопатии:
- проксимальная – патологический процесс располагается в тех частях конечностей, которые максимально близко расположены к туловищу (например, в случае с руками – это плечи);
- дистальная – поражаются мышцы конечностей, которые на максимальном расстоянии находятся от туловища (например, в случае с ногами – икроножные мышцы);
- смешанная – имеет симптомы проксимальной и дистальной форм болезни.
Первичные миопатии имеют неблагоприятный прогноз. Особенно, если первые симптомы проявились в раннем детстве. Также имеет значение вовлечение в процесс сердечной мышцы и дыхательной мускулатуры.
В этом случае прогноз заболевания ухудшается.
Наиболее благоприятный исход наблюдается при вторичных миопатиях, так как в этом случае легче достичь успех в лечении основного заболевания, вызвавшего развитие миопатии.
Симптомы
nervzdorov.ru
Миопатия начинается с появления небольшой мышечной слабости в конечностях. Быстрее наступает усталость при ходьбе или другой физической нагрузке, чем это было ранее до проявления заболевания.
Таким людям становится тяжелее преодолевать большие дистанции, поэтому зачастую приходится выполнять небольшие перерывы для отдыха, чтобы с новыми силами продолжить свой путь.
Далее к нарастающей слабости присоединяется мышечная атрофия, вследствие этого появляются деформации конечностей. Как правило, атрофические изменения наблюдаются в проксимальных (которые находятся ближе к туловищу) отделах рук и ног.
Из-за этого дистальные отделы конечностей могут казаться гипертрофированными. Это явление носит название «псевдогипертрофия». С прогрессированием мышечной слабости становится затруднительно прыгать, бегать, ходить по лестнице.
Постепенно формируется характерный вид больного при миопатии: крыловидные лопатки, опущенные плечи, выпяченный вперед живот и усиленный поясничный лордоз, за счет которого формируется так называемая «осиная» талия. Помимо этого, наблюдается «утиная» походка (передвижение сопровождается раскачиваниями в стороны).
Кроме того, миопатии могут сопровождаться поражением мимических мышц. Заподозрить вовлечение в патологический процесс мимических мышц можно в том случае, когда человек не может вытянуть губы трубочкой, надуть щеки, нахмурить лоб или улыбнуться.
Поражение круговой мышцы рта сопровождается развитием дизартрии за счет затруднения произношения гласных звуков.
Сама по себе дизартрия обозначает расстройство речи, которое выражается в затрудненном произношении некоторых слов, отдельный звуков, слогов или в их искаженном произношении.
Поражение дыхательной мускулатуры сопровождается нарушением вентиляции легких, что приводит к возникновению застойной пневмонии. Данная форма пневмонии является одной из самых тяжелых по течению, с трудом поддается лечению и быстро приводит к развитию дыхательной недостаточности.
Также имеются данные о возможности поражения сердечной мышцы. В таком случае развивается кардиомиопатия, которая приводит к развитию сердечной недостаточности.
Диагностика
nevrology-md.ru
Врач может заподозрить наличие миопатии даже на уровне первичного осмотра, так как для таких людей характерен соответствующий внешний вид.
Во-первых, наблюдается атрофия мышц (проксимальных отделов конечностей) и псевдогипертрофия дистальных отделов конечностей. Во-вторых, со временем формируется «утиная» походка.
Также для миопатии характерно опущение плеч, крыловидное отставание лопаток, выпячивание вперед живота и усиление поясничного лордоза.
Затем врач переходит к неврологическому осмотру, в ходе которого выявляет ослабление сухожильных рефлексов, снижение мышечной силы. В отличие от невропатии, при миопатии не наблюдается нарушение чувствительности.
После осмотра пациент отправляется в лабораторию для сдачи необходимого перечня анализов. Одним из них является биохимический анализ мочи, в котором ищут креатинин для подтверждения миопатии.
Наличие креатинина в моче может указывать на поражение мышц, поэтому данный показатель не лишен информативности в диагностике рассматриваемого заболевания.
Далее выполняется биохимический анализ крови, в котором оценивают уровень КФК, АЛТ, ЛДГ и других ферментов.
Помимо этого, используются элетрофизиологические методы исследования: электронейрография и электромиография.
Результаты электронейрографии дают информацию о состоянии периферического двигательного нейрона, что необходимо в случае, если присутствуют подозрения на наличие нейропатии.
Электромиография проводится для непосредственной оценки состояния мышечной ткани. При миопатии происходит изменение мышечных потенциалов (уменьшается их амплитуда и сокращается длительность).
Самым информативным методом является морфологическое исследование мышечной ткани. Это производится с помощью биопсии мышц.
Данный метод хоть и наиболее информативный, но проводится редко, лишь в тех случаях, когда предыдущие методы исследования не дали должной информации для подтверждения того или иного диагноза.
При миопатии морфологическая картина образцов мышечной ткани следующая: выявляются беспорядочно разбросанные атрофированные миофибриллы (сократительные элементы мышечных волокон), гипертрофированные мышечные волокна, а также наблюдается замещение участков мышечной ткани на соединительную или жировую.
Для оценки состояния сердечной мышцы назначается консультация кардиолога, который в свою очередь отправляет пациента на ЭКГ и УЗИ сердца. При подозрении на возникновение пневмонии, как осложнение течения миопатии, выполняется рентгенография легких.
Лечение
images.24ur.com
Для лечения первичных форм миопатии не существует препаратов, которые способны устранить генетически дефекты, послужившие развитию заболевания. Лечение в таком случае носит лишь симптоматический характер. Оно направлено на ослабление симптомов заболевания и улучшение качества жизни.
С этой целью назначаются анаболические гормоны, антихолинэстеразные препараты, витаминные комплексы. Эффект данных препаратов направлен на торможение дистрофических и атрофических изменений в мышечной ткани.
Вторичные формы миопатии лучше поддаются лечению, так как терапия направлена на устранение сопутствующих заболеваний, которые послужили причиной развития миопатии.
В случае грамотного подбора медикаментозных и немедикаметозных методов лечения наблюдается постепенное устранение симптомов, беспокоящих людей с миопатией на фоне эндокринных нарушений, хронических интоксикаций, тяжелых хронических заболеваний и т.д.
Вышеперечисленные методы лечения дополняются назначением физиотерапии. Например, используется электрофорез с неостигмином, ионофорез с кальцием и ультразвук.
Помимо этого, важно назначить массаж и ЛФК. Использование данных методов лечения направлено на замедление процесса дистрофического изменения мышечной ткани с последующей атрофией.
Также за счет постоянной двигательной активности уменьшается процесс перерождения мышечной ткани в соединительную и жировую.
Однако важно не перегрузить ослабленные мышцы, поэтому комплекс упражнений подбирается квалифицированным специалистом.
Большое внимание уделяется дыхательной гимнастике, действие которой направлено на улучшение вентиляции легких. Это необходимо для предупреждения развития осложнения в виде пневмонии, которая может возникнуть вследствие поражения дыхательной мускулатуры.
Лекарства
smartmoney.today
Цель лечения направлена на снижение атрофии мышечной ткани. В этом значительную помощь оказывают анаболические гормоны (ретаболил, неробол). Их действие выражается в стимуляции роста мышечных клеток, что необходимо для поддержания мышечной массы.
За счет этого происходит увеличение показателя силы, производительности и выносливости. Однако существует и обратная сторона приема данных препаратов. При длительном приеме могут возникнуть сопутствующие эффекты в виде облысения, увеличения роста волос на лице и теле.
Помимо этого, выделяют такое понятие, как маскулинизация, которое обозначает появление вторичных половых признаков мужского пола у женщин.
В этом случае происходит изменение телосложения по мужскому типу, огрубевает голос, нарушается менструальный цикл, изменяется эластичность кожи, появляется угревая сыпь.
Из антихолинэстеразных препаратов применяются такие лекарственные средства, как прозерин, нейромидин. Предпочтение отдается нейромидину, так как его спектр действия намного шире прозерина. Также при длительном приеме прозерина отмечаются различные неприятные побочные эффекты, поэтому в большинстве случаев препарат назначается короткими курсами.
В свою очередь нейромидин оказывает хорошее стимулирующее действие на нервно-мышечную передачу, а также увеличивает сократительную активность мышц. Кроме этого, нейромидин обладает седативным и анальгетическим (устраняет боль) действиями.
Из витаминных препаратов используются витамины группы Е, В и С, а также никотиновая кислота.
Народные средства
medaboutme.ru
Так как в большинстве случаев причиной миопатии является наследственная патология, облегчить свое состояние без медикаментозных препаратов невозможно, но с помощью самостоятельного соблюдения некоторых мер можно улучшить общее состояние.
Например, большую роль оказывает питание, поэтому важно следовать некоторым рекомендациям. Во-первых, следует внести в свой рацион больше свежих овощей и фруктов, молочные продукты, яйца, мед, орехи, крупы (особенно овсяную и гречневую). Во-вторых, важно отказаться от кофе, алкоголя, картофеля, капусты, пряных продуктов.
Для того, чтобы предотвратить ускоренную атрофию мышц, необходимо постоянно выполнять лечебную гимнастику. Но следует также помнить, что состояние мышц при миопатии далеко от состояния мышц здорового человека. Соответственно, нагрузка должна быть минимальной, чтобы не вызвать перенапряжение. С этой целью отлично подойдут занятия в бассейне.
Также не стоит забывать о важности дыхательной гимнастике. Не редкое явление при миопатии – поражение дыхательной мускулатуры, вследствие чего нарушается вентиляция легких, что в дальнейшем приводит к воспалительному процессу (развитие пневмонии). Дыхательная гимнастика направлена на улучшение газообмена в легких.
Не рекомендуется самостоятельно разрабатывать перечень упражнений, лучше обратиться к специалисту для индивидуального подбора необходимых упражнений. После обучения в рамках лечебного учреждения можно продолжить гимнастику в домашних условиях.
Информация носит справочный характер и не является руководством к действию. Не занимайтесь самолечением. При первых симптомах заболевания обратитесь к врачу.
Ваши отзывы и комментарии о лечении
Чем нас лечат: Спинраза. Против СМА и смысла

Работает ли первый в стране препарат от спинальной мышечной атрофии.
В чем Спинраза превзошла все ожидания экспертов, в чем смысл антисмысловой терапии одним из самых дорогих в мире препаратов, как гены «подменяют» друг друга, где малое число исследований и их участников простительно, а где — нет, рассказывает новый выпуск рубрики «Чем нас лечат».
В России впервые зарегистрирован препарат от спинальной мышечной атрофии — Спинраза. Раньше лечить это редкое неврологическое заболевание у нас было нечем: можно было либо получить инъекции за границей, либо ввезти и применить «по жизненным показаниям», если удастся пройти сложную процедуру оформления.
Но и сейчас стоимость лекарства достигает заоблачных высот, «превосходя даже самые смелые ожидания» западных экспертов: в США за одну инъекцию платят 125 тыс. долларов. При этом в первый год лечения их потребуется шесть, а затем их придется колоть на протяжении всей жизни. В России предельная отпускная цена не зарегистрирована, а на сайте госзакупок такой тендер еще не появился.
Давайте выясним, насколько велика вероятность улучшений после такой дорогостоящей терапии.
SMN1 и его заместитель
Спинальная мышечная атрофия (СМА) развивается из-за разрушения альфа-мотонейронов, отвечающих за сокращение скелетных мышц и находящихся в передних рогах спинного мозга. Причина этого — поломки в гене SMN1, кодирующем белок, отвечающий за выживание моторных, или двигательных, нейронов (survival motor neuron).
Мышцы не получают команду от этих нейронов, атрофируются от неиспользования, поэтому и появляются двигательные проблемы. Но у гена SMN1 есть заместитель, который образовался когда-то благодаря удвоению. В эволюционной перспективе такая подстраховка оказалась полезной: больше вероятность, что будет работать хотя бы один.
Но одно дело — «выйти за товарища на пару смен», и совсем другое — взвалить на свои плечи двойной груз обязанностей. С этим SMN2 не справляется, ведь в последовательностях двух генов есть маленькое различие: один нуклеотид («буква» ДНК) в седьмом экзоне («смысловой» вставке гена, с которой кодируется белок).
Совпало так, что при «чтении» информации с гена для синтеза белка этот нуклеотид играет скорее роль знака препинания, мешая производить белки с этого гена так же эффективно и делая готовые белки более нестабильными. Но и ген SMN2 может быть размноженным в организме.
От количества копий и успешного синтеза их белков зависит то, каким из четырех типов «классической» спинальной атрофии страдает пациент.
Спинраза стала первым в мире препаратом, бросившим вызов этой болезни. До него у пациентов была лишь надежда на заместительную терапию или симптоматические меры.
Но лекарство не только стоит очень дорого, но и вводится в позвоночный канал, в котором находится спинной мозг. Процедура непростая, трудоемкая и болезненная.
Состав препарата расскажет нам больше о том, почему форма приема именно такая и какого эффекта надеялись достичь разработчики.
Из чего же, из чего
В основе Спинразы лежит действующее вещество нусинерсен. Оно состоит из коротких цепочек нуклеотидов, которые будут связываться с определенными последовательностями нуклеотидов в промежуточной стадии производства белка — информационной, или матричной, РНК.
Такие препараты обычно называют antisense — антисмысловыми, так как они могут мешать прочтению инструкции в РНК и ее «переводу» в белок.
Но в этом случае последовательность не блокирует синтез белка, а лишь немного видоизменяет его, переводя белок в более устойчивую форму — такую же, как в норме производилась бы с гена SMN1.
Короткие цепочки нуклеотидов нусинерсена
Vaccinationist/WIkimedia Commons
Хотя изредка короткие нуклеотидные последовательности и проникают через пищеварительный тракт в кровь, это скорее исключение, чем правило.
Да и в крови антисенс-отрывки много пользы не принесут, потому что активность генов SMN1 и SMN2 важна для нас в строго определенном месте: у альфа-моторных нейронов.
Поэтому и был выбран, казалось бы, максимально неудобный и явно не подходящий для лечения на дому способ применения.
Но есть у него и плюсы: поскольку препарат не разрушается грозой большинства лекарственных молекул — цитохромами 450 — он остается активным не несколько часов, а несколько месяцев.
В плазме крови период полувыведения достигает 89 дней, а в спинномозговой жидкости — 175 дней.
Лекарственное вещество разрушают экзонуклеазы (ферменты, «режущие» ДНК) при помощи гидролиза, делая из него безобидные запчасти для новых молекул ДНК или РНК. Это значит, что повторять инъекции часто не придется.
Speranza для Spinraza
Но то, что технология работает в теории и даже в пробирке, еще не гарантирует эффективности на живых организмах — в том числе и на пациентах. В PubMed с меткой «клинические испытания» можно найти семь статей о препарате. Часть статей относится к ранним фазам клинических исследований.
Авторы одной (первая фаза, 28 человек) оценивали фармакокинетику, переносимость и безопасность препарата и не нашли проблем, а в другой (вторая фаза, 20 человек) подбирали нужную дозу.
Во втором исследовании у пациентов 77 раз случались побочные реакции, но авторы заключили, что вероятность, что они связаны с самим препаратом, мала. Скорее всего, осложнения возникали из-за самой процедуры.
В первом исследовании, по которому написали еще одну статью, без проблем тоже не обошлось: в 50% случаев побочные эффекты — головную боль, боль в спине и постлюмбарный синдром — вызвала люмбарная пункция, которую проводили для анализа того, как лекарство работает и распадается в организме.
Но для регистрации нужны испытания третьей фазы, где эффективность лекарства в двойном слепом контролируемом исследовании сравнивается с плацебо или стандартной терапией.
Двойной слепой рандомизированный плацебо-контролируемый метод — способ клинического исследования лекарств, при котором испытуемые не посвящаются в важные детали проводимого исследования.
«Двойной слепой» означает, что о том, кого чем лечат, не знают ни испытуемые, ни экспериментаторы, «рандомизированный» — что распределение по группам случайно, а плацебо используется для того, чтобы показать, что действие препарата не основано на самовнушении и что данное лекарство помогает лучше, чем таблетка без действующего вещества.
Этот метод мешает субъективному искажению результатов. Иногда группе контроля дают другой препарат с уже доказанной эффективностью, а не плацебо, чтобы показать, что препарат не просто лечит лучше, чем ничего, но и превосходит аналоги.
Американское Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (Food and Drug Administration, или FDA) одобрило препарат в 2016 году для всех типов заболевания, оценив клинические испытания на 121 ребенке с самым тяжело протекающим типом СМА, первым. Им диагностировали заболевание в возрасте до шести месяцев.
В предварительный анализ по достижении возраста шести месяцев включили 82 ребенка. У 40% детей в группе препарата развилась способность контролировать свои движения: они смогли лучше переворачиваться, ползать или стоять. В группе плацебо не было никаких изменений.
Лекарство было зарегистрировано по ускоренному алгоритму и одобрено для орфанных (редких) заболеваний, чтобы побыстрее попасть к пациентам. Вскоре одобрили применение препарата в Европе, Японии, Канаде, Бразилии и других странах. Теперь очередь дошла и до России.
В списках (не) значился
Исследование с маленькими детьми не забросили, а продолжили и опубликовали в New England Journal of Medicine. Новые оценки провели, когда пациентам исполнилось девять месяцев (в этот раз включено было 110 человек). При более длительном лечении доля отреагировавших на терапию достигла 51%, а в группе инъекции-пустышки никаких изменений к лучшему так и не наблюдалось.
В другом испытании (тоже третьей фазы) поучаствовали дети с поздним началом заболевания (после шестимесячного возраста).
Здесь улучшение быстро наступило у 41% участников из группы препарата, поэтому испытание досрочно завершили, чтобы применить лечение и к плацебо-группе.
В еще одном исследовании по следам уже проведенных оценили использование двигательного теста HINE-2, заключив, что он для испытаний подходит.
На этом клинические испытания, увы, заканчиваются. Остаются лишь обзоры того, что мы описали. Кохрейновского обзора, кстати, среди них нет — да и материала для обзоров пока маловато.
Кохрейновская библиотека — база данных международной некоммерческой организации «Кохрейновское сотрудничество», участвующей в разработке руководств Всемирной организации здравоохранения.
Название организации происходит от фамилии ее основателя — шотландского ученого-медика XX века Арчибальда Кохрейна, который отстаивал необходимость доказательной медицины и проведения грамотных клинических испытаний и написал книгу «Эффективность и действенность: случайные размышления о здравоохранении». Ученые-медики и фармацевты считают Кохрейновскую базу данных одним из самых авторитетных источников подобной информации: публикации, включенные в нее, прошли отбор по стандартам доказательной медицины и рассказывают о результатах рандомизированных двойных слепых плацебо-контролируемых клинических исследований.
Что же мы видим? Итого на основании всего двух исследований третьей фазы, в каждом из которых поучаствовало чуть больше 120 человек, препарат экстренно одобрили для всех.
Более того, оба исследования были выполнены производителем, а не независимой третьей стороной.
Это логично, поскольку препарат только разрабатывается, да и баснословная стоимость не способствует удовлетворению интереса независимых исследователей.
Indicator.Ru заключает: шансы на успех невелики, но других вариантов пока нет
В большинстве случаев лекарства всего с двумя клиническим испытаниями по сотне участников внушают мало доверия.
Но спинальная мышечная атрофия — редкое (орфанное) заболевание, и найти пациентов для тестирования лекарств достаточно трудно, поэтому небольшое их число можно простить.
Законодатели решили одобрить лекарство как можно скорее, не дожидаясь более масштабных исследований, чтобы подарить пациентам хоть какую-то надежду. В конце концов, оно достоверно продлевает жизнь и помогает начать лучше двигаться хотя бы некоторым больным СМА.
Минус препарата в том, что вылечить он сможет лишь около половины пациентов. С одной стороны, это немного (особенно учитывая стоимость лечения), с другой — это лучше, чем ничего. Было бы хорошо, если бы Минздрав включил этот препарат в специальную программу по орфанным заболеваниям, но поверить в такую перспективу пока трудно.
Радует то, что появилась «первая ласточка», и на подходе уже другие лекарства от этого заболевания. Среди них — AVXS-1016, редактирующее сломанный ген SMN1, а также различные способы коррекции работы SMN2 (включение более сильного его синтеза через усиление промотора, стабилизацию белка с гена SMN2 и так далее). Хотелось бы, чтобы они были доступны не только для миллиардеров.
Наши рекомендации нельзя приравнивать к назначению врача. Перед тем как начать принимать тот или иной препарат, обязательно посоветуйтесь со специалистом.
Источник https://indicator.ru/medicine/spinraza.htm
Миопатия — что это такое и как не проспать данный недуг?
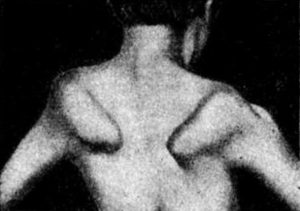
Болезни нервной системы в большинстве случаев плохо поддаются диагностике и лечению, особенно если недуг затрагивает мышечные волокна. В данной статье речь пойдет о таком заболевании как миопатия — что это такое и как бороться с данной болезнью рассмотрим подробно ниже.
Общие сведения
Итак, миопатия — это дегенеративное заболевание нервной системы, которая возникает как в результате наследственного фактора, так и вследствие провоцирующих заболеваний или травм.
Из чего состоят мышцыКстати, миопатия всегда считалась наследственной болезнью, пока не было установлена иная природа недуга.
В Неврологии имеется несколько вариантов классификации данного заболевания.
Так, по месту локализации различают:
- конечностно — поясная (поражает мышцы поясничного отдела и верхние конечности);
- лице-плече-лопаточная (поражает мышцы верхней части тела);
- гумеро — тибиальная (поражает мышцы нижних конечностей);
- глазная (поражает мышцы глаз).
Миопатия — это не что иное, как легкая форма мышечной дистрофии, которая со временем может трансформироваться в тот или иной синдром.
Так, различают:
- мышечная дистрофия Дюшенна;
- болезнь Эрба;
- болезнь Беккера;
- синдром Ландузи-Дежерина;
- дистрофия Мари-Шарко;
- дистальная миопатия (Говерса-Веландера);
- алкогольная кардиомиопатия;
- митохондриальная миопатия.
По причине возникновения различают:
- наследственная (врожденная миопатия) — первичная;
- приобретенная миопатия — вторичная.
Кстати, врожденный тип недуга еще носит название болезнь Томпсона.Также различают детскую и юношескую формы.
По глубине поражения миопатия бывает:
- проксимальная — мышцы, расположенные ближе к телу;
- дистальная — мышцы, расположенные на удалении от тела;
- смешанный тип.
Причины
Развитие миопатии любого типа возникает вследствие нарушения в функционировании митохондрий и синтеза белка в мышцах, что, в свою очередь, и приводит к дистрофии. Причин, по которым данный синтез нарушен может быть несколько.
Что касается врожденного типа то они могут быть следующими:
- генетические нарушения у матери или отца;
- заболевания, перенесенные во время беременности;
- плохая экологическая обстановка в той местности, где проживают родители больного;
- алкоголизм одного из родителей или употребление алкоголя во время беременности;
- наличие доброкачественного или злокачественного новообразования;
- частые депрессии.
В свою очередь, вторичный тип недуга может быть спровоцирован различными заболеваниями и нарушениями, в том числе:
- нарушения в работе эндокринной системы и щитовидной железы в частности;
- гормональные нарушения (тиреотоксикоз);
- системные болезни соединительной ткани (склеродермия).
Кроме того, пусковым механизмом для данного недуга могут стать:
- регулярные ОРВИ;
- пневмония бактериального характера;
- пиелонефрит;
- сальмонеллез;
- черепно-мозговые травмы;
- переломы таза;
- наркомания;
- алкоголизм;
- печеночная или сердечная недостаточность;
- авитаминоз;
- дерматомиозит;
- сахарный диабет;
- полиомиозит;
- бронхит;
- вегетососудистая дистония (ВСД);
- гипотиреоз.
Сколько может прогрессировать заболевание? Точный ответ дать сложно, так как даже врожденная форма может начать себя проявлять лишь по прошествии нескольких лет.
Дистрофия мышц происходит в связи с замещением их жировой тканью, которая, естественно, не справляется с задачей, которую мышцы выполняли ранее. Развивается слабость в конечностях и других местах человеческого тела.
В результате исследований, стало понятно, что больные миопатией испытывают проблемы с вегетативной и периферической нервной системами.
Дистрофия Дюшенна
Данное заболевание является самой распространенной и наиболее тяжелой формой болезни, а также имеет самые высокие показатели смертности. Она включает следующие симптомы:
- увеличение икроножных мышц, за счет нарастания жировой ткани;
- трудности с самостоятельным подъемом больного;
- постепенно формируется полная дистрофия всего тела;
- деформирование суставов;
- дистрофия сердечной и дыхательной мускулатуры (что может привести к летальному итогу).
Любой воспалительный процесс при подобной форме недуга может оказаться смертельным.
Дистрофия Эрба Рота
Данный вид недуга в основном развивается у малышей. Причем возможно, его развитие у грудничка, ребенка более старшего возраста или подростка.
Если речь идет о дистрофии у младенца, такое нарушение однозначно носит врожденный (наследственный) характер, так как новорожденный может приобрести данную болезнь только по наследству либо в результате генетического сбоя.
Для детей от 3 до 5 лет такой недуг также может считаться врожденным.
Ну а для более взрослых (подростков) применимо название ювенальная (юношеская) дистрофия. Причем юношеская, вовсе не означает, что девочка не сможет заболеть, женский пол подвержен данному недугу так же, как и мужской.
Основные симптомы, характерные для болезни, следующие:
- дистрофия бедер;
- атрофия спины и постепенное искривление позвоночника;
- формирование «утиной» походки и «осиной» талии;
- ослабление мышц около рта.
Говорить о мгновенном характере прогрессирования не стоит, разве что на самых ранних этапах, когда страдают совсем маленькие дети.
Дистрофия Беккера
Данная разновидность болезни обладает следующими симптомами:
- повышенная утомляемость нижних конечностей;
- изменение внешнего вида ног (вегетативные проявления);
- снижение энергообмена;
- атрофия мышц таза.
Болезнь Ландузи-Дежерина
Данное заболевание в большей степени затрагивает мышцы лица и соответственно симптоматика у нее соответствующая, в том числе:
- нарушение зрения (близорукость, дальнозоркость в несколько диоптрий);
- нарушения функционирования глазной мышцы;
- неконтролируемая фасцикуляция мышц (непроизвольное сокращение);
- слабый отклик окологубных мышц (напоминает способность двигать губами, при отеке).
Существует еще и легкая форма данной болезни — глазная миопатия, которая может давать небольшие осложнения на глаза, вызывая незначительные отклонения. Так, человек может хуже фокусировать зрение, он испытывает проблемы с их закрыванием и открыванием. Как правило, недуг такой легкой степени ничем не грозит пациенту, если его лечить.
При беременности
Беременность итак серьезный стресс для организма, и на фоне данного стресса могут возникнуть нарушения, связанные с мышечной дистрофией или миопатия.
Как правило, наиболее распространенный вариант для беременных — миопатия Беккера. Данное заболевание обычно протекает бессимптомно либо имеет несколько незначительных признаков, в том числе:
- слабость в области малого таза;
- возможны проблемы с самостоятельным вставанием с кровати или стула;
- возможно, возникновение внутренних воспалений;
- развитие таких вторичных недугов, как кифоз, лордоз, сколиоз;
- увеличение или снижение подвижности суставов.
В том случае, когда болезнь запущена и развивается поздняя стадия без вмешательства врача, возникает большой риск летального итога.
Лечить такой недуг можно только по согласованию со своим лечащим врачом, чтобы избежать негативное воздействие на плод.
Профилактика
Профилактика данного заболевания довольно специфично и включает в себя несколько рекомендаций, в том числе:
- улучшить состояние организма помогут регулярные занятия спортом (тренировки дома также подойдут);
- специальная диета (обилие молока, каши из ячменя, овса, ржи, салаты из репы и сельдерея, яблоки, больше печени, ограничение острой, соленой и жирной пищи);
- отказ от спиртного, курения;
- соблюдение режима труда и отдыха.
Прогноз
Прогноз для большинства форм недуга довольно благоприятный. Исключение составляют дистрофия Дюшенна и дистрофия Эрба Рота.
Указанные типы болезни могут закончиться летальным итогом, так как возможны нарушения сердечных и дыхательных систем (мышечные спазмы).
Тем не менее жить с миопатией можно, так как в большинстве случаев ее симптоматика поддается лечению.
Итак, миопатия является серьезным заболеванием, которое требует немедленного обращения к врачу в случае его обнаружения. Не шутите со своим здоровьем, берегите себя!
Миопатия: симптомы, принципы диагностики и лечения
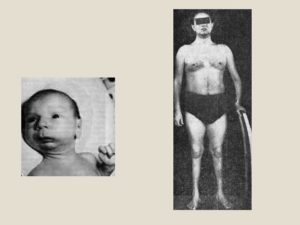
Миопатия – это разнородная группа заболеваний, в основе которых лежит первичное поражение мышечной ткани.
Другой термин миопатии – миодистрофия, который чаще используется при обозначении наследственных миопатий.
Первичное поражение мышечных клеток может происходить под действием различных этиологических факторов (наследственность, нарушение обмена веществ, бактерии). На этом факте основывается принятая классификация миопатий.
Классификация
Ведущий этиологический фактор многих видов миопатий — наследственность.
Выделяют следующие виды миопатий:
- Прогрессирующие мышечные дистрофии.
- Миопатия Дюшенна.
- Миопатия Беккера.
- Миопатия Ландузи-Дежерина.
- Миопатия Эмери-Дрейфуса.
- Конечностно-поясная форма прогрессирующей мышечной дистрофии.
- Офтальмофарингеальная форма.
- Дистальные миопатии (миопатия Миоши, миопатия Веландер и др.)
- Врожденные мышечные дистрофии и структурные миопатии.
- Метаболические миопатии (митохондриальные миопатии, эндокринные).
- Воспалительные миопатии.
В классификации указаны наиболее часто встречающиеся миодистрофии, но это не полный список.
Прогрессирующие мышечные дистрофии
Это наследственные заболевания, в основе которых лежит гибель мышечного волокна с постепенной заменой жировой тканью. Для этой группы характерно быстрое прогрессирование процесса, которое приводит к инвалидности человека.
- Миопатии Дюшенна и Беккера.
Миодистрофии имеют сходную клиническую картину. Заболевания носят рецессивный характер и передаются с Х-хромосомой, поэтому болеют только мальчики. В основе патологии лежит нарушение структуры (миопатия Беккера) или полностью отсутствие (миопатия Дюшенна) специального белка — дистрофина, который участвует в работе нейронов, мышечных волокон скелета, сердца.
Патологические изменения структурного белка приводят к некрозу мышечных клеток – атрофия. Миопатия Дюшенна встречается в несколько раз чаще миопатии Беккера. Дебют миодистрофии Дюшенна происходит в раннем возрасте (возраст от 3 до 7 лет).
Первые симптомы носят неспецифический характер, и родители на первых порах часто относят их к особенностям характера: малоподвижность по сравнению со сверстниками, пассивность в играх. Псевдогипертрофированные мышцы не вызывают подозрительности.
Со временем клиническая картина ухудшается: ребенок перестает вставать с пола без опоры, появляется утиная походка из-за слабости мышц тазового пояса. Появляется ходьба на носочках, потому что ахилловы сухожилия изменяются и препятствуют вставанию на пятки. Интеллект снижен.
Клиническое течение быстро прогрессирует и к 9-15 годам ребенок теряет способность к самостоятельному передвижению, наступает инвалидизация. При осмотре выявляются контрактуры (стягивания) в голеностопных суставах. Мышцы бедер, тазового пояса, спины, верхних отделов рук атрофируются.
Часто у детей атрофии не заметны из-за развития подкожной жировой клетчатки. Присоединяется остеопороз, дилятационная кардиомиопатия и дыхательная недостаточность.
Ребенок просыпается с чувством страха, удушья, нехватки воздуха на фоне снижения жизненной емкости легких и развития дыхательной недостаточности.
Летальный исход наступает в 20-30 лет от дыхательной или сердечной недостаточности.
Миопатия Беккера протекает более мягко. Клиническая картина в основном схожа с клиникой миопатии Дюшенна, но дебют заболевания происходит в более позднем возрасте (11-21 год). Человек теряет способность к самостоятельному передвижению в более позднем возрасте (после 20 лет). Поражение сердца встречается реже, по сравнению с миодистрофией Дюшенна. Интеллект сохранен.
- Миопатия Ландузи-Дежерина.
Заболевание поражает мышцы плечевого пояса и плеч, а также мимические мышцы лица. Дебют миодистрофии приходится на второе десятилетие жизни человека.
Первоначально появляется слабость и атрофия в мышцах плечевого пояса, которая проявляется отстоянием лопаток от спины («крыловидные» лопатки), плечевые суставы повернуты внутрь, грудная клетка уплощается в передне-заднем размере.
Постепенно в процесс вовлекаются мышцы лица: речь становится неразборчивой, опускается верхняя губа («губы тапира»), улыбка человека становится горизонтальной без поднимания уголков губ (улыбка Джоконды). У части людей атрофии поражают мышцы ног, особенно голеней.
Характерный симптом — асимметричные атрофии мышц. Псевдогипертрофии бывают не всегда. Контрактуры суставов выражены в меньшей степени по сравнению с миопатией Дюшенна.
Мышечная слабость и атрофии сочетаются с дилятационной кардиомипатией, отслоением сетчатки, снижением слуха. У некоторых пациентов двигательная активность сохраняется до конца жизни и не приводит к тяжелой инвалидизации, но другая часть пациентов приковывается к инвалидному креслу на третьем десятке жизни.
Прогрессирует медленно. Первые симптомы появляются у детей в возрасте 5-15 лет. Поражаются мышцы рук и плечевого пояса, постепенно формируются контрактуры в локтевых суставах и кистях. Одновременно атрофируются мышцы стоп, поэтому ребенок при ходьбе «шлепает» ногами.
К определенному возрасту наступает стабилизация процесса. Подъем по лестнице остается возможным без использования подручных средств. Псевдгопертрофии не характерны.
Если со стороны скелетной мускулатуры изменения не настолько выражены, чтобы приковать ребенка к инвалидному креслу, то со стороны сердца носят жизнеугрожающий характер.
Развивается дилятационная или гипертрофическая кардиомиопатия, которая нарушает работу сердца, приводит к аритмиям, блокадам, в тяжелых случая к летальному исходу. Таким пациентам устанавливают искусственный водитель ритма.
- Конечностно-поясная миодистрофия Эрба-Рота.
Встречается одинаково часто у мужчин и женщин. К особенностям клинической картины относятся развитие в 20-30 лет, инвалидизация наступает через 15 и больше лет с момента появления первых симптомов.
Мышцы плечевого пояса и тазового пояса поражаются в равной степени. При осмотре обращает на себя внимание утиная походка человека, вставание «сам по себе», «крыловидные» лопатки. Псевдогипертрофии не образуются, мышцы лица остаются интактными.
Изменения со стороны сердца не встречаются.
- Офтальмофарингеальная миодистрофия.
Проявляется опущением верхних век (птоз), поперхиванием при глотании (дисфагия) и появлением гнусавого оттенка голоса (дисфония) с последующим присоединением слабости в мышцах рук, плеч, ног и тазового пояса.
Они делятся на несколько видов в зависимости от начала болезни: с началом в грудном возрасте, с началом в детстве, с поздним дебютом (тип Веландер), тип Миоши, с накоплением десминовых включений.
https://www.youtube.com/watch?v=j7nEuUOUEso
Дистальные миодистрофии проявляются в первую очередь поражением мышц стоп и кистей. Обращает на себя внимание появление шлепающих стоп при ходьбе. Со временем может сформироваться полая стопа (увеличение свода стопы) или псевдогипертрофии мышц голеней, сколиоз.
Человека беспокоит слабость в разгибателях мышцах кистей, что создает трудности при тонко-дифференцированной работе руками. Разные подтипы дистальных миопатий прогрессирует с разной интенсивностью.
При некоторых подтипах поражение мышц распространяется выше (бедра, голени, предплечья, плечи, шея).
К редким формам прогрессирующих мышечных дистрофий относят скапулоперонеальную миодистрофию, тазово-бедренную миодистрофию Лейдена-Мебиуса, миодистрофию Мэбри и др.
Врожденные миодистрофии
Рано или поздно большинство миопатий приводят к инвалидизации больного.
Этим термином обозначают миопатии, которые возникли у ребенка сразу после рождения или в первые месяцы жизни. Диагностика заболевания основывается на следующих критериях:
- Гипотония мышц (снижение тонуса мышц) ребенка с первых дней жизни;
- Биопсийное подтверждение миопатии;
- Исключены другие заболевания со сходной клинической картиной.
С первых дней жизни у ребенка наблюдается генерализованная слабость всех мышц.
Слабость в диафрагмальных мышцах приводит к нарушению вентиляции легких и присоединению инфекции (основная причина смерти), слабость в мышцах шеи — к неспособности удерживать голову, слабость в руках и ногах – «поза лягушонка». Ребенок значительно отстает в двигательном развитии, но интеллект сохранен.
Другая отличительная особенность – контрактуры во многих суставах (локтевые, голеностопные, коленные). У части детей одновременно обнаруживаются изменения в центральной нервной системе (аномалии, демиелинизация и др.).
К врожденым миодистрофиям относят миопатию Фукуямы, врожденную миодистрофию с лейкодистрофией и цереброокулярную миодистрофию.
Причины и механизмы развития до конца не известны.
Воспалительные миопатии
По причинному фактору делятся на следующие группы:
- Идиопатические миопатии (полимиозит, дерматомиозит). В основе патологии лежит поражение мышц собственными антителами иммунной системы.
- Воспалительные (стрептококковый миозит, миозит при гриппе, токсоплазмоз и др.). вирусные, бактериальные или паразитарные агенты повреждают мышцы самостоятельно или продуктами распада.
- Воспалительные миопатии на фоне приема лекарственных средств.
Воспалительные миопатии протекают с болевым синдромом в покое и при движении мышц. При осмотре выявляется болезненность мышц, симптомы интоксикации.
Метаболические миопатии
Это заболевания имеют наследственную или приобретенную природу, в основе которых лежит нарушение обмена веществ в мышечных клетках.
Одновременно со слабостью мышц появляются другие симптомы нарушения обмена веществ, эндокринные изменения.
")
")



