Как передается миопатия по наследству
Миопатии разделяют на две большие подгруппы — первичные и приобретенные.
Первичные миопатии обусловлены генетическими нарушениями. Для них характерно постепенное прогрессирующее течение. Известные наследственные миопатии — ювенильная миопатия Эрба, миопатия Дюшенна, плече-лопаточно-лицевая миопатия Ландузи-Дежерина, болезнь Помпе (для которой доступна патогенетическая терапия)1.
https://www..com/watch?v=ytcreatorsru
К первичным относятся дистальные формы (дистальная мышечная дистрофия).
Это группа редких прогрессирующих генетических нарушений, при которых происходит истощение (атрофия) и слабость мышц, удаленных от центра тела, — мышц рук и ног.
К ним относится дистальная миопатия Миоши, дистальная мышечная дистрофия с вакуолями «в оправе» (Нонака), Веландера, Лейна, тибиальная мышечная дистрофия Удда, другие2.
Первичные формы связаны с одной или более генетической мутацией, которые ответственны за появление дефектов в белках, обеспечивающих мышечный тонус и сокращение, а также некоторых ферментов. Например, одна из самых распространенных наследственных форм, миопатия Немалина, обусловлена мутациями в 10 генах.
Дефектный ген может наследоваться рецессивно или доминантно, иногда он наследуется сцеплено с Х-хромосомой.
Приобретенные миопатии разделяют на воспалительные (дерматомиозит, полимиозит), инфекционные, метаболическое, эндокринные, лекарственные, токсические. Они развиваются на фоне сопутствующих заболеваний и патологических состояний, например:
- Приема лекарственных препаратов (глюкокортикостероиды);
- Приема алкоголя;
- Поступления в организм токсинов (токсическая форма);
- Эндокринных расстройств (нарушение функции щитовидной железы и др.);
- Авитаминозов;
- Сердечной недостаточности;
- Нарушения работы печени;
- Злокачественных заболеваний, др.
Причины заболевания
Заболевание, в зависимости от типа, имеет различные причины.
Воспалительные формы развиваются на фоне аутоиммунных реакций — нарушения работы иммунной системы, из-за которого организм «атакует» собственные ткани, органы. Некоторые воспалительные формы связаны с системной красной волчанкой, ревматоидным артритом, узелковым полиартериитом4.
Инфекционными причинами могут становиться вирусы, бактерии и другие возбудители, например:
- Трихинеллы;
- Токсоплазмы;
- ВИЧ-инфекция;
- Вирусы Коксаки;
- Вирусы гриппа;
- Бактерии рода Borrelia-возбудители болезни Лайма;
- Золотистые стафилококки.
Предлагаем ознакомиться: Алименты платят до
Лекарственные миопатии могут развиваться на фоне приема ряда препаратов:
- Глюкокортикостероиды;
- Ловастатин, другие статины — препараты, снижающие уровень холестерина;
- Колхицин — противоподагрическое средство;
- Амиодарон — препарат, применяемый при сердечных аритмиях.
Как проявляется болезнь?
При первичных формах первые признаки заболевания проявляются обычно в детском или юношеском возрасте. Типичный симптом, по которому можно заподозрить заболевание у ребенка, — мышечная слабость.
Этот признак характерен для всех форм. Обычно слабость носит симметричный характер, то есть наблюдается в симметричных мышцах.
На начальных стадиях слабость может быть незначительной, но постепенно ее выраженность увеличивается.
С прогрессированием заболевания снижается качество жизни: для больного становятся сложными, трудновыполнимыми даже привычные нагрузки. Появляются трудности при ходьбе, подъеме по лестнице.
Из-за дистрофии мышц может нарушаться осанка, появляться искривление позвоночника, например, поясничный лордоз, кифоз, сколиоз, прогрессирующие со временем.
При этом голова и живот больного выпячиваются вперед, плечи опускаются, формируются крыловидные лопатки3.
При поражении проксимальных, расположенных ближе к центру тела мышц появляются сложности с подъемом со стула, выходом из ванны, подъем по лестнице, расчесыванием волос или бритьем.
Наблюдается «утиная» походка — пациент передвигается, раскачиваясь в стороны.
При слабости в кистях у человека появляются трудности при выполнении высокодифференцированной работы (письмо, игра на музыкальных инструментах, токарное дело и др.).
Слабость стоп проявляется формированием полой стопы, шлепающей походкой.При некоторых формах, например, миопатии Немалина, иногда при болезни Помпе, появляется слабость дыхательных мышц, что приводит к повышению риска легочных инфекций. К тому же ухудшается снабжение кислородом, из-за которого могут страдать мозг, сердце, другие органы4.
Наряду с мышечной слабостью снижаются сухожильные рефлексы, возникают мышечные спазмы или сокращения, ограничение подвижности сустава — контрактура3.
Отдельные виды миопатий: клиническая картина и особенности
Дистальная миопатия Миоши — самая распространенная дистальная форма, хотя точное число больных пока установить невозможно. Имеет аутосомный тип наследования. Основные клинические признаки — прогрессирующая мышечная слабость, атрофия отдельных мышц. Чаще заболевание проявляется в подростковом возрасте, изредка — после 40 лет. В первую очередь поражаются стопы, голени.
Предлагаем ознакомиться: Как вступить в наследство после смерти брата?
https://www..com/watch?v=ytcopyrightru
Характерные жалобы в начале манифестации заболевания — трудности при вставании на пальцы, слабость при ходьбе на носках.
Вскоре икроножные мышцы начинают атрофироваться, иногда очень значительно уменьшаясь в объеме всего за несколько дней. Постепенно слабость появляется в мышцах задней поверхности бедра, ягодиц, запястий, пальцев.
Болезнь прогрессирует медленно, инвалидизация наступает обычно через несколько десятков лет после появления первых признаков5.
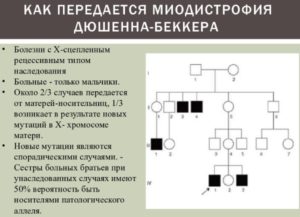
Мышечная дистрофия Дюшенна — одна из наиболее распространенных наследственных форм. Наследуется по рецессивному, сцепленному с Х-хромосомой типу. Развивается только у мальчиков.
Симптомы появляются в возрасте 2-5 лет.
Клинические признаки — быстрая утомляемость, трудности при подъеме по лестнице, частые падения, прогрессирующая слабость мышц, атрофия — мышечная ткань заменяется фиброзом или жировой тканью.
Болезнь Помпе — редкое прогрессирующее нервно-мышечное заболевание, передается по аутосомно-рецессивному типу.
Связана с дефицитом лизосомального фермента альфа глюкозидазы, вследствие чего в клетках накапливается гликоген, поэтому заболевание относят к лизосомным болезням накопления.
Заболевание характеризуется прогрессирующей дегенерацией скелетной, дыхательной, и, в основном, у младенцев — сердечной мышечной ткани.
Болезнь Помпе может развиваться в любом возрасте, в том числе и у взрослых. Основные проявления — мышечная слабость, атрофия мышц. Это приводит к нарушению походки, сложностям при ходьбе, подъеме по лестнице, гиперлордозу. Дыхательные симптомы проявляются одышкой при незначительных нагрузках, частыми инфекциями верхних дыхательных путей, легких.
Дистальная мышечная дистрофия Нонака наследуется по аутосомному рецессивному типу. Симптомы начинаются в 20-30 лет с поражения передних большеберцовых мышц.
Через несколько лет нарушается функция стопы, затем поражаются мышцы бедер и подвздошно-поясничные мышцы. Больным становится сложно подниматься по лестнице, вставать со стула.
Вскоре присоединяется слабость сгибателей шеи, плечевого пояса, кистей. Через 13 лет после начала больные не могут самостоятельно передвигаться8.
Как диагностировать миопатию?
Чтобы установить диагноз, одной клинической картины недостаточно, однако появление характерных признаков заболевания должно стать поводом для глубокого обследования пациента.
Предлагаем ознакомиться: Можно ли носить крестик доставшийся по наследству
При подозрении на врожденную миопатию необходимо проведение молекулярно-генетических методов исследования. Без них установить окончательный диагноз невозможно.
Кроме того, обследование включает3:
- Биохимический анализ крови, который помогает оценить уровень ферментов, АЛТ, АСТ, КФК (креатинфосфокиназы), лактатдегидрогеназы и др.;
- Электромиографические, электронейромиографические методы исследования — первые помогают оценить потенциалы, которые возникают в скелетных мышцах при возбуждении мышечного волокна, а вторые определить функциональное состояние мышц и периферических нервов;
- Биопсия мышечного волокна, с ее помощью можно выявить атрофические, разрушительные процессы;
- Иммунобиохимические, иммуногистохимические исследования, необходимые при подозрении на аутоиммунную патологию.
Лечение миопатий
В настоящее время патогенетической терапии для большинства миопатий не существует, однако разрабатываются методы генной и клеточной терапии, а для миопатий, связанных с болезнями накопления, — терапия, позволяющая замещать недостаточность фермента в организме. Для некоторых заболеваний лечение уже доступно. К ним относятся болезнь Помпе.
Помимо патогенетического лечения проводятся симтоматическая терапия. Она включает различные классы препаратов, усиливающих процессы обмена веществ, увеличивающих мышечную силу, поддерживающих нормальную работу нервной системы, головного мозга, сердца, органов пищеварения3.
Немедикаментозное лечение включает целый ряд терапевтических методик, направленных на улучшение качества жизни, социализации. К ним относится лечебная физкультура, физиотерапия, психологическая реабилитация, массаж, другие.
https://www..com/watch?v=https:accounts.google.comServiceLogin
Прогноз наследственных миопатий может быть различным. Крайне важна своевременная диагностика.
Что нам известно о миопатии Эрба

Миопатия Эрба возникает у детей и юношей, обуславливается генными мутациями. У развивающегося плода такую болезнь могут провоцировать разные факторы.
Такой болезни подвержены потомки здоровых граждан и малыши из неблагополучных семейств. Патология диагностируется у детей от 14 до 18 лет, но может проявляться в раннем возрасте около 3 лет. Только у мальчиков проявляется Миопатия Эрба.
Классификация миопатий
С учетом того, какие мышцы подвержены повреждениям, преобразуется классификация болезни. Миопатия Дюшена считается наиболее распространенной разновидностью и отличается наивысшей степенью сложности.
Эта патология имеет наследственный характер в большинстве примеров, развивается стремительно. Проблема с работой дистрофина, регулирующего прочность мембран, является основной причиной возникновения болезни.
Зачастую такая патология возникает только у мужчин. Девушки в большинстве примеров выступают в роли переносчиков. Развивается такая миопатия с трехлетнего возраста, отличается отчетливой симптоматикой, о которой свидетельствует заболевание.
Если в столь юном возрасте малыш еще способен совершать какие-то телодвижения, то к 12 он уже не может шевелиться. Наиболее подвержены икроножные мышцы на ногах, при которых они утолщаются. Возникает сколиоз и другие возможные повреждения.
При миопатии Беккера возникает сердечная недостаточность. Это усугубляет общее положение. Преобразование можно проследить на первичных этапах при повреждении миокарда.
Это все можно определить после выполнения ЭКГ или ЭхоКГ. Если пренебречь терапией этой патологии, может развиваться слабость в мышцах и проблемы с дыханием.
Такая симптоматика может привести к летальному исходу.
Для миопатии Беккера различают 2 способа диагностики:
- Генодиагностика.
- Анализ дистрофина в мышечных тканях.
Зачастую ко второму варианту прибегают при подозрениях на эту болезнь, поэтому диагноз может быть подтвержден или опровергнут.
терапевтическая методика направлена на предотвращение патологии. ЛФК может поспособствовать уменьшению мышечных преобразований, используются приспособления, облегчающие передвижение. К хирургическим процедурам прибегают в сложных ситуациях. Миопатия при этом угрожает жизнедеятельности людей.
Главной причиной заболевания является генетическое преобразование, развивающееся в процессе внутриутробного развития. Миопатия Эрба может передаваться по наследству. Наиболее отчетливым признаком является слабость м мышечных тканях. Наблюдается атрофирование и прекращение их возможного развития.
Поэтому через какое-то время они высыхат, исчезает возможность свободно передвигаться. При этом пациенты не испытывают болезненные симптомы, присутствует только слабость. Она не исчезает даже после продолжительного сна, через какое-то время начинает усугубляться.
Причины болезни и возможность появления осложнений
Миопатия Эрба-Рота считается первичной патологий, возникающей после какого-то периода из-за плохой наследственности или генетических преобразований. Главным фактором является проблема с генофондом плода при беременности, которая происходит из-за патологии или курении, употреблении спиртного при беременности.
Развивающемуся плоду наносится колоссальный вред. У человека развивается гиперлордоз, признаки крыловидных лопаток, лицевые нервы портят визуальные характеристики.
К причинам нарушения генофонда относятся:
- Наследственность.
- Работа во вредных для здоровья условиях.
- Взаимодействие с химическими реагентами.
- Постоянное употребление антибиотиков.
- Поздняя беременность.
- Постоянные нервные напряжения.
Развивающаяся мышечная дистрофия при появлении осложнений может быть смертельно опасной. Состояние может усугубиться такими способами:
- Паралич.
- Грыжа.
- Миотонические дистрофии.
- Чрезмерное разветвление нервных волокон.
- Нехватка кислорода.
- Затруднения с передвижением.
- Пневмония.
Чтобы избавиться от подобных осложнений или отсрочить их возникновение, нужно выполнять советы специалистов.
Симптомы
Слабость и атрофия мышечных тканей считается главными признаками миопатии Эрба. Боли пациенты не испытывают, постоянная слабость не проходит после продолжительного отдыха. На первых стадиях заболевания возникают незначителные кратковременные улучшения после сна и отдыха. Спустя какое-то время слабость возобновляется.
Начинают проявляться крыловидные лопатки. Походка становится переливающейся. Грудь и живот выдвигаются вперед. Основной отличительной особенностью является гипомимическая физиономия с отчетливо выведенными вперед губами.
Преобразованию подлежит часть субнервального аппарата. Пациенты с миопатией Эрба отличаются тонкой талией. Быстро развиваются контрактуры, поэтому сокращаются мышечные ткани в области стоп и голеней.
Пациент начинает хромать, опирается на большие пальцы. Концентрация жирных кислот в организме повышается. В кровеносной системе увеличивается уровень калия.
Пациентам тяжело подниматься по ступенькам или вставать со стула.
При попытке подняться из лежачего положения пациент старается упереться руками и проделать несколько этапов для подъема. После этого мышечная дистрофия переходит в атрофию, мускулатура на руках и туловище истончается.
Процесс заболевания продолжительный, потому пациента, ощущая слабость, могут ходить самостоятельно до 45 лет и дольше.
Диагностика
Диагноз патологии определяется без трудностей. При диагностике дистрофии Эрба учитывается возрастная категория, наследственный фактор, скорости развития патологии. При осмотре у невролога удается определить ухудшение рефлекторной деятельности до начала выпадения, мышечный тонус усугубляется, присутствуют контрактуры суставов.
Вопреки возможным заблуждениям, фасцикулярных подергиваний мышечных тканей не происходит. При регистрации биотоков в мышечных тканях уменьшается амплитуда, но не частотность разрядов. По ЭНМГ выявляется снижение продолжительности потенциалов действия, полифазность.
Часто выявляется преобразование деятельности креатиникиназы, АСТ и прочих ферментов. Может преобразоваться состав электролитов. Достоверным является диагноз при выполнении гистологического обследования мышечных тканей.
Форма и габариты мышечных волокон преобразуются, ткани начинают восстанавливаться, повышается их объем. Между волокнами возникает жир или соединительная ткань.
Волокна не распределяются отдельными почками, как при нейронных миопатиях.
Способы лечения
До беременности девушкам нужно как можно меньше взаимодействовать с химическими реагентами и употреблять спиртное. В процессе вынашивания плода нежелательно пить спиртное или курить.
Употреблять антибиотики тоже нельзя. Когда у детей проявляется миопатия Эрба, специалисты рекомендуют посещать массажистов хотя бы 2 раза в месяц. Больше выполнять упражнения ЛФК, прогуливаться на воздухе, плавать, побольше физической активности.
Все главные терапевтические методики врачи советуют проводить в стационаре.
Могут прописать такие медикаменты:
- Какинтон.
- Церебролизин.
- АТФ.
- Витамины.
Возможные осложнения
Юношеская миопатия Эрба отличается возможными осложнениями, от которых люди зачастую умирают. Среди них зачастую возникают:
- Проблемы с дыхательной функцией.
- Полная утрата способности двигаться самостоятельно.
- Пневмония.
- Межпозвоночные грыжи.
- Искривление.
- Парезы.
- Параличи.
Предотвратить такие осложнения не получится, но можно отсрочить их возникновение
Сегодня нет методики, позволяющей избавиться от юношеской миопатии Эрба. Врачи советуют пациентам выполнять упражнения по ЛФК, ходить в бассейн, гулять на улице, быть более активным. Каждые несколько месяцев нужно делать массаж, но назначенные медицинские процедуры предпочтительно проводить в стационаре.
Прогноз при различных мышечных дистрофиях зачастую негативный. Патология со временем усугубляется, охватывает мышечные ткани. Со временем подвижность пациента ухудшается. При этом патология не приводит к летальному исходу. Смерть наступает чаще из-за пролежней, инфекционных процессов.
Не передать по наследству

Генетика за последние 10–20 лет шагнула далеко вперед, и сегодня ей известно около пяти тысяч наследственных заболеваний. Современная наука располагает большими возможностями для выявления наследственных и врожденных патологий у ребенка еще до его появления на свет.
Если будущие родители займут в вопросе планирования беременности более ответственную позицию – будут активно обращаться к специалистам, соглашаться на генетические исследования и дородовую диагностику, постоянно наблюдаться – то процент рождения неизлечимо больных детей с тяжелыми пороками развития станет намного меньше.
В идеале сегодня каждая супружеская пара, планирующая детей (или партнеры, желающие иметь совместного ребенка), должна проходить медико-генетическую консультацию у специалистов – врачей-генетиков в женских консультациях, платных клиниках или НИИ генетики РАМН. Есть в стране и специализированные медико-генетические консультации.
Ухудшающаяся экология, постоянные стрессы и прочие «прелести» жизни в современных мегаполисах не способствуют рождению здорового потомства и лишь увеличивают число врожденных патологий у детей.
У любой супружеской пары риск рождения ребенка с пороками развития составляет 6%, поскольку каждый человек является носителем генных изменений и мутаций, передающихся из поколения в поколение, и багаж этот нарастает как снежный ком.
Каждый из нас несет в своем хромосомном наборе в среднем 10–12 дефектных генов, и даже если вы и все ваши отцы, деды и прадеды до седьмого колена были здоровы, нет никакой гарантии, что именно с вашим наследником природа не сыграет злую шутку и дефект не проявит себя в виде какой-то патологии. Наиболее перспективными считаются новейшие методы диагностики – ДНК-диагностика и молекулярно-генетический анализ.
Кто в группе риска?
Но в большинстве случаев будущие родители пренебрегают визитом к генетику, надеясь на авось. Врачи оставляют принятие решения об обследовании на усмотрение мужчины и женщины, но далеко не всегда.
Есть немало случаев, когда генетическая консультация назначается по медицинским показаниям: если у будущих родителей осложненный анамнез, то есть у отца или матери есть родственники, дети или сами они страдают каким-то наследственным заболеванием; если у женщины были 1–2 выкидыша; если будущие родители – дальние родственники; если женщина старше 35 лет; если мужчина старше 50 лет.
Если все-таки будущие родители по каким-то соображениям решили не консультироваться с генетиками, от дородовых обследований беременной женщине все же отказываться не стоит.
Ведь исследования делаются на 14–16-й неделях беременности, и если у плода выявятся дефекты развития, у родителей будет время для принятия решения на основе полученной информации – прерывать беременность или нет.
Обследовав будущую маму, врачи смогут сказать, насколько серьезная патология у ребенка, как она отразится на его здоровье и вообще, жизнеспособным он родится или нет. Медики лишь сообщают информацию, но последнее слово всегда остается за женщиной и мужчиной.
Методы дородовой диагностики наследственных болезней, давно и широко используемые на Западе, все активнее применяются и в России.
Так, в соответствии с приказами Минздравсоцразвития РФ, сегодня каждая беременная женщина должна трижды пройти обследование на генетические дефекты плода, которое состоит из УЗИ, компьютерной томографии и исследования белково-сывороточных факторов.
Эти виды диагностики позволяют выявить около 70% врожденных патологий, в том числе такие хромосомные заболевания, как болезнь Дауна или болезнь Эдвардса, предопределяющяя умственную неполноценность ребенка, пороки развития сердца, нервной трубки, головного мозга, аномальное развитие рук и ног.
Что касается синдрома Дауна, то в настоящее время в России ведется постоянный скрининг по выявлению патологии – обследуют всех беременных россиянок, причем женщин старше 35 лет – дважды в обязательном порядке (известно, что чем старше беременная, тем выше вероятность болезни Дауна у ребенка).
Дело в том, что синдром Дауна – одна из самых распространенных наследственных патологий: даунята рождаются с частотой 1 на 800 здоровых детей. Немало и других серьезных хронических заболеваний, которые передаются по наследству от матери. Например, инсулинозависимый сахарный диабет (первого типа) или бронхиальная астма.
Анализы первого дня
Младенцев также обследуют на наследственные и врожденные заболевания – не явные, а уже скрытые, которые невозможно выявить с помощью ультразвука и других дородовых диагностических методов. Например, в США всех новорожденных обследуют на 8 наследственных болезней, в России – на 5–6.
Так, все малыши без исключения еще в роддоме сегодня проходят анализы на фенилкетонурию, врожденный гипотериоз, диагностику на адреногенитальный синдром, галактоземию и муковисцидоз.
У каждого маленького человечка на пятый день жизни берут кровь и отправляют ее на генетический анализ, чтобы определить, нет ли повреждений в генах.
Фенилкетонурия – не что иное, как олигофрения, умственная отсталость, развития которой при своевременной диагностике сегодня можно избежать. Фенилаланин – аминокислота, которая есть во всех белковых продуктах и которая в норме расщепляется и выводится из организма.
Но если процесс метаболизма нарушен, аминокислота и токсины накапливаются в организме, что приводит к тяжелому поражению ЦНС, изменениям в мозгу и слабоумию.
Чтобы этого не случилось, детей с непереносимостью фенилаланина уже с трех недель жизни переводят на специальную диету, и они нормально развиваются, растут, ходят в обычные детские сады, школы и вырастают здоровыми, полноценными людьми, а не идиотами.
Казалось бы, всего лишь диета – дети до 12–18 лет получают продукты, содержащие белок, освобожденный от пагубной аминокислоты, – но если ее не соблюдать, ребенок обречен на умственную отсталость. Каждый год в России выявляют около 150 детей, не переносящих фенилаланин, и порядка 450 детей, организм которых (вернее, щитовидная железа) не вырабатывает гормон тироксин. При своевременно выявленной врожденной патологии всем им врачи могут помочь.
Вирус-убийца: найти и обезвредить
Есть еще одна проблема, с которой сталкиваются будущие мамы и на которую не стоит закрывать глаза, – хронические инфекции половых органов. Еще на этапе планирования беременности и женщина, и ее партнер должны сдать комплексный анализ на инфекции.
В него входят цитомегаловирус, токсоплазмоз, хламидиоз, герпетическая и урогенитальные инфекции. Какие-то из них, если нет обострения, не опасны для ребенка, а какие-то надо обязательно сначала лечить, а потом уже беременеть. Например, хламидиоз или герпес.
Особенно опасна последняя: если женщина заражается герпесом во время беременности, очень велика степень рождения ребенка с очень серьезными проблемами со здоровьем или что беременность закончится выкидышем.
Герпетическая инфекция существует двух типов: первый поражает губы, носогубный треугольник, глаза, шею и руки, второй – гениталии и ягодицы.
Именно второй тип очень опасен для беременной и ее ребенка: вирус может стать причиной тяжелейших врожденных заболеваний – эпилепсии, слепоты, глухоты, детского церебрального паралича. «Герпес убивает мозг», – говорят врачи, и если будущая мать заражается генитальным герпесом, нередко ставится вопрос о прерывании беременности – слишком уж велик риск рождения неполноценного младенца.
КОМУ НЕОБХОДИМА МЕДИКО-ГЕНЕТИЧЕСКАЯ КОНСУЛЬТАЦИЯ НА ЭТАПЕ ПЛАНИРОВАНИЯ БЕРЕМЕННОСТИ:
– если в роду встречались наследственные заболевания: гемофилия, сахарный диабет, миопатия Дюшена – тяжелое поражение мышечной системы и другие.
Генетики определят, является ли кто-либо из будущих родителей носителем дефектного гена, вызывающего болезнь, и могут ли они передать его ребенку;– если в семье уже есть ребенок с врожденными пороками развития, генетическая консультация необходима для того, чтобы выяснить, является ли врожденное заболевание наследственным;– если супруги состоят в родственном браке, например троюродные брат и сестра и у них есть общие прадеды и прапрадеды. В этом случае мужчина и женщина могут быть носителями одних и тех же дефектных генов;– если у кого-либо из родственников была задержка умственного или физического развития, не связанная с внешней причиной – травмой, перенесенным инфекционным заболеванием;
– если мужчина или женщина уже в зрелом возрасте, в стареющих клетках при образовании зародыша хромосомы не всегда расходятся правильно, что влечет врожденные уродства и патологии.
Наследственные миопатии и миодистрофии, их клинические проявления и прогноз

Все наследственные заболевания нервно-мышечной системы являются, на первый взгляд, достаточно редкими: один случай встречается на 3, а то и на 50 тысяч человек. Но если взять число жителей в большом городе с населением в несколько миллионов человек, то окажется, что количество только одних пациентов с симптомами миодистрофии Дюшенна в «городе-двухмилионнике» приближается к 600 случаев.
Поэтому в каждом крупном городе должен быть создан если не центр, то хотя бы кабинет нервно-мышечных заболеваний, с возможностью оказания неотложной помощи, например, при миастенических кризах.
Что такое наследственные нервно-мышечные болезни, и как они проявляются?
Эти заболевания имеют прогрессирующее течение, и их можно разделить на две различные группы:
- когда первично поражены мышцы (миопатии), в них находятся дефектные ферменты, аномалии строения белка. Такие болезни именуют также миодистрофиями;
- когда первично поражены периферические, двигательные нейроны, которые расположены в передних рогах спинного мозга. Эти заболевания называют спинальными амиотрофиями в случае поражения тел нейронов, и невральными амиотрофиями – если тела нейронов здоровы, но наблюдается нарушение нервно – мышечной проводимости в аксоне, или нерве.
Наконец, в отдельной статье рассмотрена миастения, которая является удивительным заболеванием: и нервы, и мышцы здоровы, но страдает синапс – месть соединения концевой пластинки нерва с мышечным волокном.
Наследственные миопатии
Примером болезней, протекающих с симптомами наследственных миопатий – детская псевдогипертрофическая мышечная дистрофия Дюшенна, поздняя дистрофия Беккера, плечелопаточная дистрофия Ландузи-Дежерина, и другие, более редкие формы: лопаточно – перонеальная форма Давиденкова, ювенильная дистрофия Эрба, офтальмоплегическая форма миопатии, дистальная миопатия Веландера, а также ряд врожденных и непрогрессирующих форм. Рассмотрим симптомы наиболее часто встречающихся миопатий.
Детская миодистрофия Дюшенна
Является злокачественной дистрофией, и встречается довольно часто – 1 случай на 3,5 тысячи, и только у мальчиков. Частота не зависит от расы, географии, социального положения. Болезнь проявляется прогрессированием мышечной слабости. Кроме этого, миопатия приобретает признаки мышечной гипотрофии, а затем – и атрофии.
Болезнь начинается рано, мышечная слабость возникает вначале в районе мышц таза, затем плеч, в возрасте ребенка до 3 лет.
Поздняя псевдогипертрофическая миодистрофия Беккера
По клинике это заболевание протекает так же, как и болезнь Дюшенна. Но при этом оно начинается после 10-летнего возраста, возникает в 8 раз реже, чем предыдущее заболевание.
Прогноз при миодистрофии Беккера намного более благоприятный, вследствие очень медленного прогрессирования болезни и длительного сохранения функции движения у пациентов. Иными словами, пациенты способны передвигаться самостоятельно до возраста 35-40 лет. Нарушений интеллекта у них не бывает.
Заболевание также кодируется в виде рецессивного типа, сцепленного с полом, и для диагностики миодистрофей Беккера и Дюшенна можно применять одни и те же пробы ДНК.
Миопатия Ландузи – Дежерина
Это более редкий вид миопатии. Ее еще называют плечелопаточно-лицевой, по той причине, что поражаются мышцы лица и плечевого пояса. Мимика снижена, веки полностью не смыкаются, круговая мышца рта гипертрофирована, возникает симптом «губ тапира».
Возникает гипотрофия, атрофия и парезы в проксимальных отделах рук, появляется такой симптом, как «крыловидные лопатки». При далеко зашедшем течении возникает поражение мышц ног и тазового пояса.
В таком случае пациент с миопатией Ландузи – Дежерина может быть оформлен на получение пособия по инвалидности.
Это заболевание связано с тем, что фермент КФК (креатинфосфокиназа) не может превращать креатин в креатинфосфат, связывая его с АТФ. В результате мышца не получает высокоэнергетического соединения, которое может использоваться при повышенной мышечной работе. В результате накапливается КФК в мышцах, происходит их атрофия, а в поздних случаях – денервация.
Лечение при миопатии Ландузи – Дежерина симптоматическое. Прогноз – для жизни благоприятный, для качества жизни – возможно инвалидизация, начиная с возраста в 30-40 лет.
При всех наследственных миопатиях специфического лечения не разработано.
Применение гормонов приводит к замедлению прогрессирования поражения мышц, при выраженной дистрофии мышц показано хирургическое лечение (например, операция артродеза) при привычном вывихе болтающегося сустава.
Выводы
Все наследственные миопатии относятся к генетическим редким заболеваниям, и при помощи пренатального скрининга можно установить точно, болеет ли будущий малыш той или иной формой миопатии.
В том случае, если известно точно «генеалогическое дерево» и история заболевания, то врач генетик может и заранее, до зачатия точно предсказать вероятность появления дефектного гена на свет, и рекомендовать воздержаться от зачатия, особенно гетерозиготным носителям гена.
В том случае, если после зачатия рождается девочка, то она больной не будет, но может нести дефектный ген. Поэтому консультация у генетика – это очень важный этап в планировании семьи у подобных пациентов.
Миопатия
Миопатии представляют группу нервно-мышечных заболеваний, которые проявляются утомляемостью, слабостью мышц, снижением мышечного тонуса, атрофией мышц.
Миопатии, в зависимости от причинного фактора, разделяются на прогрессирующую наследственную мышечную дистрофию, эндокринные Миопатии (заболевание желез внутренней секреции) и метаболические Миопатии (нарушение обмена веществ).
Прогрессирующая наследственная мышечная дистрофия или Миопатия характеризуется атрофией мышц за счет разрушения мышечных клеток в связи с недостатком специального белка дистрофика (примечание: белок называется ДИСТРОФИН), который укрепляет структуру мышечных волокон.
Этот белок вырабатывается под контролем специального клеточного гена, что расположен на 6-й хромосоме человека, и при дефекте этого гена наступает постепенное разрушение оболочек мышечных клеток с последующим омертвением мышечных волокон.
Этот дефектный ген передается по наследству, если в роду был брак между родственниками (примечание: Изменения гена в 30% случаев происходит в результате мутации. Брак между родственниками — ни при чём! Болезнь передаётся по наследству с вероятностью 50%, если один из родителей ребенка болен). Он связан с женской половой хромосомой и передается, как правило, сыновьям, хотя сами женщины могут и не болеть.
Формы дистрофии
Атрофии подвергаются мышцы плечевого пояса рук, спины, тазового пояса и ног. В зависимости от локализации болезни, возраста, тяжести заболевания выделяют различные формы мышечных дистрофий.
Так, юношеская форма дистрофии Эрба-Рота возникает в возрасте 10-20-ти лет, когда незаметно появляется атрофия мышц плечевого пояса и рук, а затем — тазового пояса и ног. Во время ходьбы больной переваливается с выпяченным вперед животом и отодвинутой назад грудной клеткой.
Чтобы встать из положения лежа, больной поворачивается на бок и, опираясь руками на бедра, постепенно поднимает свое туловище. Болезнь медленно прогрессирует.
Детская форма мышечной дистрофии Дюшена начинается в возрасте 3-5-ти лет с атрофии мышц таза, бедер с одновременным утолщением икроножных мышц голени (ложное утолщение). Постепенно атрофируются мышцы плечевого пояса и рук.
У детей вначале нарушается походка, а затем возникает сложность в передвижении. У многих нарушается сердечный ритм за счет увеличения размеров сердца. Прогрессирование заболевания или его злокачественное течение в связи с ранним обездвиживанием конечностей приводит к печальному исходу. Болеют, в основном, мальчики (1 на 3000 рожденных).
(примечание: Болеют и мужчины и женщины в равной степени. Только болезнь Дюшена проявляется у мальчиков. Девочки являются носителями этого гена.)
Но бывает и доброкачественное течение мышечных дистрофий (миодистрофия Беккера), когда заболевание проявляется медленно, особенно у низкорослых детей. Многие годы они сохраняют удовлетворительное физическое состояние и только присоединение различных острых заболеваний и травм приводит их к обездвиженности, истощению с плохим исходом.
Выделяется плече-лопаточно-лицевая форма миодистрофии, называемая дистрофия Ландузи-Дежерина, которая может быть в возрасте от 6-ти до 52-х лет (чаще в 10-15 лет) и характеризуется поражением мышц лица с постепенной последующей атрофией мышц плечевого пояса, туловища и конечностей.
Ранними признаками болезни являются плохо смыкающиеся и незакрывающиеся веки, полиостью не смыкающиеся губы что создает нечеткую речь и невозможность надуть щеки. Заболевание протекает медленно.
Долгое время больной может передвигаться и сохранять трудоспособность, а затем через 15-25 лет постепенно атрофируются мышцы тазового пояса ног, что затрудняет передвижение.
В отличие от вышеуказанных миодистрофии, редко встречается окулярная или офтальмическая Миопатия с ограничением движения глазных яблок, опущением век, нарушением глотания, слабостью мышц плечевого пояса, а также дистальная Миопатия, при которой отмечается медленно прогрессирующая атрофия мышц рук и ног. Заболевание в среднем начинается в возрасте 5-15-ти лет.
Вторичные дистрофии
Выделяется также группа вторичных прогрессирующих мышечных дистрофий, которые возникают в связи с поражением нервов: невральная, спинальная миодистрофии, называемые еще амиотрофиями.
- К невральной относится амиотрофия Шарко-Мари, которая характеризуется постепенной атрофией мелких мышц стоп, затем атрофируются мышцы голеней и нижней части бедер, а мышцы средней и верхней частей бедер не изменяются и бедро представляет форму бутылки с горлышком, опрокинутым вниз. Затем постепенно атрофируются мышцы кистей рук и предплечий.
Не поражаются мышцы туловища, плечевого пояса и лица. Заболевание возникает в возрасте 18-25 лет, медленно прогрессирует и стабилизируется.
- Врожденная спинальная мышечная атрофия Кугельберга-Веландера характеризуется постепенной атрофией мышц рук, ног, задержкой психического и физического развития, деформацией позвоночника.
Болезнь проявляется в возрасте 8-10-ти лет и медленно прогрессирует.
- Прогрессирующая амиотрофия Арана-Дюшена начинается в возрасте 25-50-ти лет и проявляется атрофией мышц кистей. Затем постепенно атрофируются остальные мышцы рук, потом ног туловища, в т.ч. межреберных мышц, что вызывает дыхательные нарушения, от которых наступает смерть.
- Врожденная амиотония (снижение тонуса мышц) Оппенгейма характеризуется слабостью мышц в связи с их недоразвитием, и мышечная дистрофия их является вторичной. У новорожденных она не прогрессирует, но попадание респираторных инфекций может вызвать воспаление, и смерть наступает на первом году жизни С возрастом двигательная функция мышц улучшается.
- Спинальная амиотрофия Вердинга-Гоффмана начинается в раннем детском возрасте (в первый год жизни) и проявляется атрофией мышц конечностей, спины, а потом всего туловища, быстро прогрессирует, исход — неблагоприятный Бывают и другие редкие разновидности прогрессирующих мышечных дистрофий.
Лечение дистрофии
Лечение мышечных дистрофий направлено на замедление дистрофических (разрушающих) процессов в мышцах и даже их прекращение. Однако радикального лечения пока не найдено.
Хотя надежда есть на генную терапию, которая начинает медленно внедряться в медицинскую практику. В Израиле применяются новейшие лекарства для лечения мышечной дистрофии различных типов, одобренные Мин.
Здравом и разрешенные для применения на территории Израиля.
Другие виды миопатии
- Эндокринные Миопатии бывают при тиреотоксикозе (токсический зоб), когда к основным его признакам: увеличение щитовидной железы, пучеглазие и тахикардия (учащенное сердцебиение) присоединяются мышечная слабость в области мышц плеч, шеи, бедер, дрожание рук и всего тела, похудение, повышенная возбудимость, плохой сон.
У больных с гипотиреозом (снижение функции щитовидной железы) к основным признакам заболевания (вялость, сонливость, запоры, сухость кожи, снижение памяти, температуры тела) присоединяется Миопатия в виде болезненных и уплотненных мышц с судорогами (синдром Гоффмана).
- При аддисоновой болезни (недостаток гормонов коры надпочечников), кроме пигментации кожи и слизистых в виде бурой окраски, снижения артериального давления, уменьшения размеров сердца, похудения, желудочно-кишечных расстройств (понос и рвота), также присоединяется миопатия в виде резкой мышечной слабости, быстрой утомляемости, мышечных болей и судорог.
- При первичном альдостеронизме (синдром Конна) отмечается увеличение синтеза альдостерона в клубочковом слое коры надпочечников. При этом повышается выведение калия в кишечнике, потовых и слюнных железах, что создает щелочную реакцию, высокую артериальную гипертензию и Миопатию в виде мышечной слабости, болевого спазма мышц ног, а иногда приступообразного паралича мышц ног, который длится от нескольких часов до нескольких дней. Снять болевой приступ можно внутривенным введением хлорида калия или панангина.
- При синдроме Иценко-Кушинга (нарушение функции гипоталамо-гипофизарно-надпочечниковой системы) наряду с выраженным отложением жира в области лица, шеи, живота, сухости кожи, расстройством половой функции, повышением артериального давления, сахарным диабетом, язвой желудка и др. отмечается миопатическая дистрофия — истончение мышц рук и ног. Для лечения эндокринных Миопатий применяют различные гормональные препараты и др.
- Метаболические Миопатии (нарушение обмена веществ) бывают при первичном амилоидозе мышц, когда амилоид или сложный мукополисахарид откладывается в мышцах, и последние приобретают каменистую плотность и болезненность Болезнь начинается с детства. Такая же метаболическая миопатия наблюдается при гликогенозах. Лечение направлено на восстановление белково-углеводного обмена.
Михаил Сенявский,врач,г. Могилев.
Из газеты «Народный доктор» август 2000 г. №16(86)
")
")



