Миопатия у детей — лечение, причины и симптомы миопатии
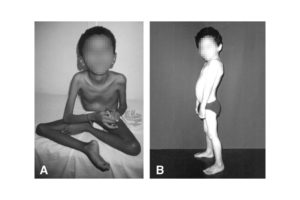
Миопатия – это дегенеративное заболевание мышечных волокон, обусловленное генетическими факторами. В основе болезни лежит прогрессирующая атрофия мускулатуры, приводящая к первичной мышечной дистрофии.
Помимо того что болезнь передается по наследству, ее можно получить в результате инфекции или травмы (вторичная миопатия). Но чаще всего заболевание встречается у представителей одной семьи.
Первичная миопатия развивается в детском возрасте. Под воздействием неблагоприятных факторов (физическое переутомление, острые или хронические инфекционные болезни, отравления) симптомы патологии усугубляются.
Вторичные миопатии регистрируются намного реже, они являются следствием дисфункций вегетативной и эндокринной систем.
Помимо этого различают миопатии:
- проксимальную – поражаются мышцы конечностей, находящиеся ближе к туловищу;
- дистальную – страдают мышцы, удаленные от тела;
- смешанный тип – проксимальная и дистальная одновременно.
Миопатия: виды
В группу миопатий принято включать хронические, постепенно прогрессирующие заболевания, связанные с поражением, исчезновением мышечных волокон, заменой их жировыми или соединительными тканями. Четкой классификации на сегодня не существует.
Многие исследователи выделяют патологии по области преимущественного поражения, например, лице-лопаточно-плечевую, конечностно-поясную. Другие говорят о миопатиях в зависимости от характера причин – наследственных или приобретенных. Разделяют патологии по тому, что преимущественно поражается – белки или ферменты.
Принято также выделять отдельные болезни:
- Мышечная дистрофия Дюшенна. Болезнь встречается в 0,03% случаев. Развивается у мальчиков до 5 лет. Это одна из самых тяжелых форм миопатий. В среднем к 15 годам ребенок теряет возможность двигаться и самостоятельно себя обслуживать. Патология поражает сначала мышцы нижних конечностей, затем – верхних. В некоторых случаях развивается умственная отсталость.
- Ювенильная мышечная дистрофия Эрба-Рота. Заболевание регистрируется обычно в подростковом и юношеском возрасте до 20 лет. Поражаются мышцы таза и нижних конечностей. Больного отличает походка, напоминающая утку, тонкая талия. Отмечаются крыловидные лопатки. Обследование выявляет ухудшение сухожильных рефлексов. Из положения лежа больные встают с помощью рук.
- Плече-лопаточно-лицевая форма Ландузи-Дежерина. Миопатию этой формы отличает поражение мышц лица, плечевого пояса, лопаток. Страдают мышцы-разгибатели пальцев. Обнаруживается изменение формы груди. Заболевание впервые диагностируется в возрасте от 10 до 20 лет. В отличие от многих миопатий эта форма развивается медленно и имеет относительно благоприятный прогноз.
- Дистальная миопатия (тип Веландера). Появляется после 20 лет. Связана с уменьшением объема мышечной ткани в голеностопном отделе, коленях, предплечьях, кистях рук. Инвалидизация может наступить через 10 лет, а то и позже.
- Поздняя дистрофия Беккера. Обнаруживается у детей от 5 лет и молодых людей до 20 лет. Проявляется слабостью мышц, высокой утомляемостью, заменой мускульной ткани жировой. Сначала поражаются мышцы таза, бедер, голени, затем болезнь поражает руки. Интеллект остается сохранным.
Понятие и характеристика
Что это такое? Миопатия представляет собой заболевание, характеризующееся прогрессирующими дистрофическими изменениями мышечной ткани и скелетной мускулатуры.
В медицинской практике данным термином объединяется группа болезней со схожими морфологическими признаками.
В большинстве случаев патология развивается на фоне генетических факторов, но проявиться может в любом возрасте.
Миопатия всегда сопровождается снижением тонуса мышц, уменьшением объема активных движений и атрофией определенных участков мышечной ткани.
Симптомы
Наиболее ярко признаки патологии проявляются в миопатии Дюшенна. Симптомы у детей проявляются в возрасте от полутора лет. Страдают сначала мышцы ног. Ребенку становится трудно ходить, он быстро устает, падает, не может подняться по лестнице. Падения и усталость приводят к тому, что он испытывает страх и старается передвигаться сам как можно меньше. Походка начинает напоминать утиную.
Постепенно ребенок может подняться, используя только руки, но постепенно слабеют и они. Со временем поражаются мышцы груди, сердца, дыхательных органов.
Синдром Дюшенна характеризуется постепенным исчезновением мышц, однако внешне это бывает незаметно. Наоборот, кажется, что они становятся больше. Происходит это из-за замены мышечной ткани жировой.
Заболевание приводит к нарушению скелета. Уменьшается объем движения суставов, искривляется позвоночник, деформируется стопа, пальцы.
Поражение мышц сердца ведет к появлению одышки, аритмии, нестабильности давления. Дыхание становится поверхностным.
Проявления опасного заболевания
Заболевание обладает целым рядом клинических симптомов, но основной признак миопатии – патологическая слабость определенной группы мышц.
Медики выделяют следующие симптомы миопатии:
Формы миопатии и виды: классификация, таблица

Миопатия – прогрессирующее дегенеративное нервно-мышечное заболевание, при котором происходит нарушение обмена веществ, влекущее поражение мышечных волокон с последующей атрофией. Формы миопатии и ее виды могут быть различны.
Механизм развития миопатии
В результате нарушения обмена веществ, происходит разрастание жировой и соединительной тканей, миофибриллы не могут сокращаться, что влечет ограниченность движений. Конечная стадия, мышечная атрофия, ведет к полной обездвиженности.
Виды миопатий
В зависимости от этиологии миопатии бывают наследственные и приобретенные.
Наследственные – генетически обусловленные нарушения функционирования клеточных структур, отвечающих за обмен веществ в мышцах. В обычных условиях болезнь может не проявляться, пока ее не спровоцируют неблагоприятные факторы – инфекции, интоксикации, травмы, физические нагрузки.
Приобретенные формируются как следствие других заболеваний:
- эндокринных – гипер-, гипотиреза, болезни Иценко — Кушинга, гиперпаратиреоза;
- хронических интоксикаций – профессиональных вредностей, наркомании, алкоголизма, токсикомании;
- авитаминозов;
- нарушения пищеварения (мальабсорбция);
- хронической сердечной, почечной, печеночной недостаточности;
- злокачественных новообразований.
Формы миопатии
Классификация миопатий по формам представлена в таблице
| Виды | Формы | |||||
| Наследственные | Ювенильная/юношеская форма Эрба | Псевдо-гипертрофическая форма Дюшена | Плече — лопаточно — лицевая форма Ландузи — Дежерина | Дистальная | Окуло-фарингеальная | Скапуло-перонеальная |
| Врожденные | Болезнь центрального стержня | Немалиновая | Миотубулярная | Диспропорция ти-пов миофибрилл | Мерозин — негативная | |
| Воспалительные | Инфекционные | Вирусные | Паразитарные | Идиопатические | ||
| Метаболические | Связанные с нарушением липидного обмена | Связанные с нарушением обмена гликогена | Связанные с нарушением метаболизма пуринов | Митохондриальные | ||
| Мембранные | Миотония Томпсона | Болезнь Беккера | ||||
| Токсические | На фоне алкоголизма | На фоне наркомании | В результате хронической интоксикации | Обусловденные профессиональ-ными вредностями |
Рассмотрим некоторые формы миопатий подробнее.
Наследственные
Ювенильнаяконечностно—пояснаямиопатияЭрба – начинается в 20 – 30 лет с поражения мышц таза, бедер, постепенно распространяясь на весь организм. Мышцы лица не поражаются. Чем раньше началось заболевание, тем неблагоприятнее прогноз. Молодые люди быстро теряют обездвиженность, в зрелом возрасте процесс тянется годами.
ПсевдогипертрофическаямиопатияДюшена – болезни подвержены мальчики 3, реже – 5-10 лет. Начинается с мышечного аппарата таза и конечностей.
Сопровождается искривлениями позвоночника – кифозом, сколиозом, гиперлордозом. Ребенок отстает в физическом и умственном развитии.
Рано формируются контрактуры суставов, поражаются дыхательные и сердечные мышцы (у 90% пациентов отмечается кардиомиопатия) . Эта форма часто заканчивается летально.
Плече—лопаточно—лицевая– характерна для возраста 10-20 лет. В отличие от предыдущих форм, начинается с поражения мимических мышц. Постепенно в процесс вовлекаются мышцы верхних конечностей и груди. Эта форма не затрагивает мышцы таза. Течение вялотекущее. Работоспособность сохранена, угроза для жизни отсутствует.
Скапулоперонеальная— атрофии и нарушениям чувствительности подвержены мышцы верхних и нижних конечностей.
Окулофарингеальная– характерна для людей среднего и старшего возраста. Проявляется в сочетании атрофии глазодвигательных (глазная форма) и языкоглоточных мышц. Начинается с двустороннего птоза, позже присоединяются нарушения глотания.
Дистальная– начинается с кистей и стоп с последующим распространением на предплечья и голени. Течение медленное.
Врожденные
Обусловлены изменением генов, ответственных за синтез того или иного белка, находящегося в составе мышечной ткани.
Клинически проявляются нарастающей мышечной слабостью с частым присоединением дыхательной и сердечной недостаточности, составляющих угрозу для жизни. Если осложнений нет, врожденные миопатии протекают вяло, без прогрессирования.
Отличаются они по типу наследования, локализации мутантных генов и виду дефектных белков. О врожденной миопатии у детей.
Мерозин— негативная(«классическаявосточная») – характеризуется врожденной мутацией гена, обеспечивающего синтез белка мерозина. Наблюдается сочетание атрофии проксимальных отделов конечностей с дыхательной недостаточностью.
Воспалительные
Развиваются в результате инфекционно — вирусных заболеваний. Если возбудитель не определен – это идиопатическая миопатия.
Метаболические формируются на фоне обменных и клеточных нарушений.
Мембранные
Возникают вследствие дефектов в мембранах мышечных волокон. Сопровождаются миотоническим синдромом – замедленным расслаблением мышц после их активного сокращения.
МиотонияТомпсона – с аутосомно-доминантным типом наследования, болезнь Беккера — с аутосомно — рецессивным типом наследования (встречается реже). Характерно раннее начало, ребенок не может совершать быстрых движений, затруднено глотание, дефекты речи.
Токсические
Развиваются на фоне интоксикаций.
Диагноз «Недифференцированная форма миопатии» ставят в случае, если по этиологии и клинике она не подходит ни под один известный вид.
Выделяют ряд редких форм заболевания, как врожденных, так и приобретенных. Приобретенные всегда протекают благоприятнее.
Диагностика того или иного вида и формы заболевания ставится на основании данных:
- осмотра;
- электромиографии;
- электронейрографии;
- биохимического анализа крови;
- биопсии мышц.
Общие признаки миопатий
Несмотря на разнообразие видов, заболевание имеет общие признаки.
Первые симптомы – мышечная слабость в конечностях, быстрая утомляемость после физической нагрузки.
Постепенно, в течение нескольких лет слабость нарастает, появляется деформация костей и суставов. Больному трудно ходить, подниматься по лестнице, вставать со стула, пола, постели.
Он не может совершать активные движения. Характерны вставание «лесенкой» (поэтапно) и специфическая «утиная» походка (раскачивание во время ходьбы).
Постепенно атрофия мышц становится тотальной, возникают контрактуры суставов.
Параллельно может наблюдаться атрофия мимических мышц, что выражается в невозможности надуть щеки, свистеть, нахмуриться, улыбнуться.
На поздних стадиях вследствие деформации костно-мышечной системы, происходит нарушение работы внутренних органов :
- со стороны дыхательной системы – застойная пневмония, дыхательная недостаточность;
- со стороны сердечно –сосудистой системы – кардиомиопатия, сердечная недостаточность.
Лечение и прогноз
Лечение наследственных форм – симптоматическое, так как этиология до конца не изучена. Цель терапии – восстановить обмен веществ, улучшить трофику мышц. При присоединении проблем со стороны внутренних органов назначают средства, корректирующие работу сердечно-сосудистой, дыхательной систем, нормализующие когнитивные функции.
Терапия приобретенных видов – патогенетическая: антибиотикотерапия при инфекционных и вирусных заболеваниях, дезинтоксикационная при интоксикациях, гормональная при эндокринных нарушениях.
Помимо лекарственных средств, назначают физиотерапию, ЛФК, массаж. Лечение проводят курсами по 1,5 месяца три раза в год.
Прогноз зависит от формы заболевания, возраста пациента, в котором оно дебютировало, присоединения осложнений. Миопатии, начавшиеся в раннем возрасте, наиболее опасны.
Алина Вейц, врач-психоневролог, кандидат психологических наук, специально для Mirmam.pro
Мышечная дистрофия приговор или испытание на прочность?
В современной неврологии существует огромное количество заболеваний, характер возникновений которых не поддаются рациональному объяснению со стороны специалистов. К таким болезням можно отнести группу таких недугов, как мышечная дистрофия. Всего разновидностей данной болезни девять, но обо всем по порядку…
Общие сведения
Мышечная дистрофия — это хроническое наследственное заболевание в результате развития которого происходит поражение мышечной системы человека. Пораженные мышцы перестают нормально функционировать, истончаются в размерах, а в месте их расположения в организме постепенно нарастает жировая прослойка.
Разновидности мышечной дистрофии
В современной неврологии данный недуг классифицируется на девять различных болезней. Классификация заболевания связана с:
- локализацией мышечных нарушений;
- характеристиками болезни;
- агрессивностью развития;
- возрастом.
Так, мышечная дистрофия бывает:
- Дюшенна;
- миотоническая (болезнь Штейнерта);
- Беккера;
- Эрба Рота;
- юношеская форма дистрофии Эрба-Рота;
- плече — лопаточная лицевая форма (Ландузи-Дежерина);
- алкогольная миопатия;
- дистальная форма;
- миодистрофия Эмери —Дрейфуса.
Дистрофия Дюшенна
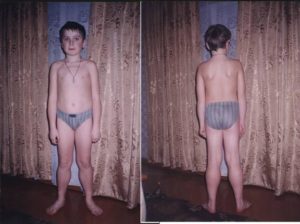
Наиболее известная форма прогрессирующая дистрофия Дюшенна (псевдогипертрофическая дистрофия, мерозин — негативная врожденная дистрофия). Данная разновидность недуга проявляется в детском возрасте начиная с двух до пяти лет.
В первую очередь от данного недуга страдают мышцы нижних конечностей, которые несмотря на малоподвижный образ жизни у маленьких пациентов, постепенно увеличиваются в размерах.
Данная особенность связана с нарастанием жировой ткани вместо мышц.
Слева здоровый ребенок, справа больной
Постепенно, по мере прогрессирования болезни она переходит в верхнюю часть и поражает мышцы верхних конечностей. Как правило, уже к двенадцатилетнему возрасту маленький пациент перестает полностью двигаться. Летальность у данного заболевания очень высока примерно 85–90% больных не доживают до 20-летнего возраста.
В группе риска находятся представители мужского пола, так как девочек данный недуг практически не затрагивает.
Болезнь Штейнерта
Врожденная дистрофия Штейнера не зря имеет название миотоническая, так как в результате прогрессирования болезни происходит слишком медленное расслабление мышц после их сокращения (такое явление называется миотонией).
Данное заболевание, в отличие от предыдущего распространено у взрослых, в возрасте от 20 до 40 лет. Имеют место быть и случаи прогрессирования болезни у детей, как правило, в младенческом возрасте, однако, это скорее исключение, чем правило.
На лицо признаки миотонии (приоткрытый рот и веки)
Гендерной зависимости болезнь не имеет и в равной степени от нее страдают как мужчины, так и женщины. Отмечается, что при недуге проявляется слабость мимических мышц лица, а также конечностей. Прогрессирование, в отличие от болезни Дюшенна происходит медленно.
Отличительная черта заболевания, возможность поражения не только мышц конечностей, но и внутренних мышц (сердечная мышца), что в свою очередь, представляет большую опасностью для жизни человека.
Болезнь Беккера
Данная форма болезни также долго прогрессирует, и пациент длительное время чувствует себя хорошо. Обострение недуга может произойти на фоне полученных травм или различных заболеваний нервной системы, которые своим течением ускорят развитие недуга.
В группе риска находятся люди низкого роста.
Болезнь Эрба-Рота и юношеская ее форма
Данное аутосомно — рецессивное заболевание развивается у пациентов после 20 лет, а юношеская форма у детей и подростков 11—13-летнего возраста. Прогрессирование болезни происходит по восходящему варианту, то есть сначала поражаются мышцы нижних конечностей, и постепенно происходит восхождение недуга к верхним конечностям.
Отличительной чертой недуга является наличие выступающих лопаток, которые по мере прогрессирования выступают более явно и очевидно.
Во время ходьбы отмечается переваливание пациента, выпячивание живота и задвигание грудной клетки.
Болезнь Ландузи-Дежерина
Плече-лопаточно-лицевая форма данного заболевания является наиболее безразборным видом, так как поражает людей в возрасте от пяти до 55 лет. Для данного недуга характерно очень длительное развитие, пациент может сохранять трудоспособность до 25 лет жизни с данной болезнью.
Отличительными симптомами является поражение лицевых мышц, в результате чего у больного могут возникнуть проблемы с четкостью произношения, в связи с неполным смыканием губ. Кроме того, отмечается неполное смыкание век.
По мере развития у больного атрофируются лицевые мышцы, мышцы плеч, конечности и туловища.
Данная форма не зависит от генетической мутации.
Алкогольная миопатия
Данный вид болезни также несвязан с генными мутациями и причина его возникновения одна — злоупотребление алкоголем. У больных могут отмечаться болевые синдромы в конечностях, связанные именно с поражением мышц.
Различают острую и хроническую алкогольную миопатию.
Дистальная форма
Дистальная форма мышечной дистрофии является доброкачественным вариантом прогрессирующей дистрофии. Как правило, данное заболевание трудно дифференцировать от невральной амиатрофии Мари-Шарко. Для разделения этих двух заболеваний требуется проведение электроэнцефалограмму головы, которая и дает понимание с какой болезнью предстоит иметь дело.
К основным симптомам недуга относят атрофию мышц конечностей с их последующим исхуданием. Возможны парезы стоп, кистей рук и т. п.
Миодистрофия Эмери —Дрейфуса
Данный тип болезни первоначально, вообще, не был выделен в качестве отдельного заболевания, так как по своей симптоматике был схож с дистрофией Дюшенна. Однако, в дальнейшем в результате длительного исследования было установлено, что болезнь Эмери —Дрейфуса обладает индивидуальной симптоматикой.
Классификация по местам поражения
Болезнь относят в разряд редких. В группе риска находятся люди до 30 лет, как правило, юный возраст. Есть свидетельства о проявлении симптомов и после 30 лет, но они единичны.
Основное отличие данного вида болезни — это проблемы с мышцами сердца, которые в итоге могут стать причиной смерти. Кардиомиопатия при данном недуге не единственное отличие. Кроме проблем с сердцем, у пациентов наблюдают стандартные для дистрофии Дюшенна признаки, но в более щадящем режиме развития.
Причины
Негативная составляющая болезней нервной системы заключается в том, что они тяжело поддаются изучению. По этой причине не до конца известно о факторах, провоцирующих развитие той или иной формы мышечной дистрофии.
Основная причина, для формирования большинства подвидов данной болезни является генная мутация, в частности гена, который отвечает за синтез белка.
К примеру, болезнь Дюшенна имеет прямое отношение к мутации половой Х хромосомы. Основной переносчик нехорошего гена выступают девушки, которые несмотря на его наличие в собственной ДНК не страдают от данной болезни.
Что касается миотонической формы, то виновник ее возникновения ген, расположенный в 19 хромосоме.
Основные симптомы
Наличие большого количество различных подвидов у данного недуга указывает на различия в симптоматике, однако у болезни есть общие симптомы, которые в себя включают:
- сниженный болевой порог или полное его отсутствие;
- снижение тонуса мышц;
- наличие болевых ощущений в пораженных участках;
- планомерная атрофия скелетных мышц;
- изменения в манере ходьбы;
- невозможность устойчиво стоять на ногах и передвигаться, вследствие чего больной часто спотыкается и падает;
- хроническая усталость;
- изменение мышц в размерах как в большую, так и в меньшую сторону;
- постепенное утрачивание навыков у детей, которые им только получилось приобрести и развить.Некоторые сигналы дистрофии
Диагностика мышечной дистрофии
Диагностические мероприятия при подобном недуге обширны, так как существует большое количество болезней, от которых необходимо дифференцировать болезнь.
На первоначальном этапе доктор обязательно изучит анамнез больного, уточнит о симптоматике, режиме дня и т. п. Эти данные требуются для составления плана проведения последующих диагностических мер, которые могут в себя включать:
- электромиография;
- соскоб мышечной ткани для анализа;
- генетическое тестирование;
- анализ крови и мочи;
- дополнительная консультация ортопеда, гинеколога и терапевта.
Стоит отметить, что чем позже проявилась болезнь, тем лучше для больного, так как ранние симптомы в большинстве случаев заканчиваются смертью.
Лечение
Лечение мышечной дистрофии процесс сложный и затяжной, однако, в настоящее время не создано лекарство, которое полностью исцеляет больного. Все мероприятия направлены на облегчение жизни больного и восстановления некоторых утраченных способностей.
Для затормаживания развития болезни назначают следующие препараты:
- кортикостероиды;
- витамины В1;
- аденозинтрифосфат (АТФ).
Кроме того, для замедления процесса развития используют фетальные стволовые клетки, которые замедляют процесс дистрофии.
Помимо того, в качестве профилактических мероприятий назначают:
- массаж;
- физиотерапию;
- дыхательную гимнастику.
Помимо стандартных вариантов лечения, важно постоянно руководствоваться тремя главными составляющими в процессе жизнедеятельности:
- Адекватная физическая активность.
- Своевременная психологическая поддержка.
- Соблюдение диеты.
Физическая активность
Отсутствие желания у человека бороться с недугом оказывает негативное воздействие на организм. Судите сами, пассивность, нежелание двигаться угнетает и без того пораженную мышечную систему. Мышцам необходимо давать нагрузку, так как без нагрузки дистрофичные процессы начинают происходить быстрее, тем самым прогрессируя в более быстром темпе.
Умеренная физическая активность, использование поддерживающих приспособлений будут отличным подспорьем в борьбе с недугом.
При наличии болей в мышцах отлично подойдет плавание, йога, упражнения на растяжку.
Психологическая поддержка
Психологическая поддержка окружения важна для больного человека. А если недуг такой серьезный, как этот тем более. Для кого-то будет достаточно обычной поддержки со стороны друзей и родных, а кому-то может потребоваться квалифицированная психологическая помощь.
Важно дать понять такому человеку, что он не остался один на один со своей проблемой. Он должен понимать, что ему есть к кому обратиться, есть люди, которые сопереживают и поддерживают его.
Диета
Что касается режима питания и соблюдения диеты, существует расхожее мнение, что соблюдение противовоспалительной диеты может замедлить прогрессирование недуга. Данная диета снижает воспаление, уровень глюкозы, выводит токсины из организма и питает его полезными веществами.
Суть такой диеты в следующем:
- Отказ от продуктов, содержащих «плохие» жиры и замена их на «хорошие», введение в рацион ненасыщенных жиров, которые содержатся в оливковом, льняном, кунжутном масле, авокадо.
- Применение в пищу мяса и рыбы, при производстве которых не были использованы антибиотики или гормоны.
- Полное удаление из рациона рафинированного сахара и глютена.
- Употребление в пищу следующих продуктов — китайская капуста, брокколи, сельдерей, ананас, лосось, свекла, кукумария, имбирь, куркума и других продуктов, обладающих противовоспалительными свойствами.
- Молочные продукты допустимы только на основе овечьего и козьего молока.
- Допустимо употребление травяных чаев, лимонада, кваса, морсов и натуральных соков.
Профилактика
Так как мышечную дистрофию довольно тяжело предсказать и обнаружить на раннем этапе, профилактические мероприятия сводятся к двум простым рекомендациям:
Обязательное обследование женщины на этапе планирования беременности на предмет наличия мутаций в генах
В случае если до беременности, по каким-либо причинам не было сделано генетического обследования, уже в период беременности проводится тест на определение у плода мутаций в Х хромосоме.
Прогноз и осложнения
В зависимости от вида болезни прогноз может быть разным, и тем не менее можно выделить несколько возможных осложнений, возникающих при данном недуге.
- нарушения сердечной деятельности
- искривление позвоночника
- снижение интеллектуальных способностей больного
- снижение двигательной активности
- нарушения дыхательной системы
- летальный итог
Итак, мышечная дистрофия довольно опасное и неизлечимое заболевание, поэтому будущим родителям необходимо с глубокой ответственностью относиться к планированию беременности. Берегите себя и своих будущих детей!
Миопатия Дюшена. Что это такое, симптомы у детей, взрослых, проноз, лечение, тип наследования
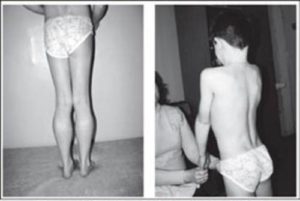
Что это такое? Миопатия представляет собой заболевание, характеризующееся прогрессирующими дистрофическими изменениями мышечной ткани и скелетной мускулатуры.
В медицинской практике данным термином объединяется группа болезней со схожими морфологическими признаками.
В большинстве случаев патология развивается на фоне генетических факторов, но проявиться может в любом возрасте.
Миопатия всегда сопровождается снижением тонуса мышц, уменьшением объема активных движений и атрофией определенных участков мышечной ткани.
Немалиновая миопатия
Второе название данного заболевания — врожденная непрогрессирующая нитеобразная миопатия. Наследственность в основном передается по аутосомно-доминантному типу, но встречается также рецессивный и спорадический. Возможен летальный исход вследствие дыхательной недостаточности в раннем младенческом возрасте.
Наблюдается сильно выраженные патологии скелета. Развитие болезни может происходить в той или иной степени, а может не прогрессировать вовсе.
В отдельных случаях больные вынуждены передвигаться с помощью сидячей каталки, в других — страдают от дыхательной недостаточности.
При диагностировании проводится гистологическое исследование, которое выявляет в мышцах неподобные или палочкоподобныене малиновые тельца. ЭМГ обычно утверждает диагноз миопатии.
Причины возникновения
Основной причиной развития миопатии у детей является наследственная предрасположенность. Этиологические факторы, способствующие развитию данного недуга, в медицинской практике остаются неизученными.
Специалисты выделяют несколько таких причин, но в некоторых случаях миопатия может стать осложнением определенного заболевания в сочетании с генетическими предпосылками, которые на протяжении длительного времени не были выявлены у ребенка.
Причинами миопатии у детей могут быть следующие факторы:
- генетическая предрасположенность;
- серьезные нарушения обмена веществ в организме;
- прогрессирование патологических процессов в мышечных тканях;
- регулярные и чрезмерные переутомления детского организма;
- последствия травм, сопровождающихся повреждением мышечной ткани;
- последствия заболеваний соединительной ткани;
- нарушения симпатической иннервации;
- патологические изменения в щитовидной железе;
- нарушение синтеза креатина и креатинина в организме;
- последствия инфекционных заболеваний;
- истощения организма различной этиологии.
Как делать массаж при гипертонусе мышц у ребенка? Читайте об этом здесь.
Как проявляется болезнь?
При первичных формах первые признаки заболевания проявляются обычно в детском или юношеском возрасте. Типичный симптом, по которому можно заподозрить заболевание у ребенка, — мышечная слабость.
Этот признак характерен для всех форм. Обычно слабость носит симметричный характер, то есть наблюдается в симметричных мышцах.
На начальных стадиях слабость может быть незначительной, но постепенно ее выраженность увеличивается.
С прогрессированием заболевания снижается качество жизни: для больного становятся сложными, трудновыполнимыми даже привычные нагрузки. Появляются трудности при ходьбе, подъеме по лестнице.
Из-за дистрофии мышц может нарушаться осанка, появляться искривление позвоночника, например, поясничный лордоз, кифоз, сколиоз, прогрессирующие со временем.
При этом голова и живот больного выпячиваются вперед, плечи опускаются, формируются крыловидные лопатки3.
При поражении проксимальных, расположенных ближе к центру тела мышц появляются сложности с подъемом со стула, выходом из ванны, подъем по лестнице, расчесыванием волос или бритьем.
Наблюдается «утиная» походка — пациент передвигается, раскачиваясь в стороны.
При слабости в кистях у человека появляются трудности при выполнении высокодифференцированной работы (письмо, игра на музыкальных инструментах, токарное дело и др.).
Слабость стоп проявляется формированием полой стопы, шлепающей походкой.При некоторых формах, например, миопатии Немалина, иногда при болезни Помпе, появляется слабость дыхательных мышц, что приводит к повышению риска легочных инфекций. К тому же ухудшается снабжение кислородом, из-за которого могут страдать мозг, сердце, другие органы4.
Наряду с мышечной слабостью снижаются сухожильные рефлексы, возникают мышечные спазмы или сокращения, ограничение подвижности сустава — контрактура3.
Наследуется аутосомно-доминантным способом с неполной пенетрантностью (но встречаются и спорадические случаи наследственности). Данная форма врожденной миопатии характеризуется патологией проксимальных мышц конечностей, но больные способны приобрести некоторые двигательные навыки.
В младенческом возрасте наблюдается задержка двигательного развития и гипотония, но диагностировать данное заболевание можно только в более позднем возрасте при изменениях скелета и выраженной мышечной слабости.
При этом наблюдаются патологии скелета: деформация стоп, кифосколиоз, дислокация бедер, грудь сапожника.
Чаще всего, больные имеют хрупкую фигуру и невысокий рост.
При диагностике заболевания проводят биоптат мышц, который показывает наличие множественных или единичных прерывистых зон, которые лишены ферментов окисления, в некоторых мышечных волокнах.
Проведение других лабораторных анализов может показать норму. Пациенты с болезнью центрального стержня склонны к развитию злокачественной гипертермии.
Классификация и формы
Миопатия может быть первичной или вторичной. В первом случае патология развивается в качестве самостоятельного заболевания, во втором — становится последствием прогрессирования других болезней, оказывающих негативное воздействие на соединительную и мышечную ткань ребенка.
В медицинской практике первичная миопатия подразделяется на три отдельные группы — врожденная (симптоматика проявляется в первые месяцы жизни), ранняя детская (патология свойственна детям от пяти до десяти лет) и юношеская форма (проявляется в подростковом возрасте).
Мерозин-дефицитная врождённая мышечная дистрофия
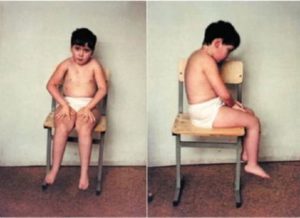
Обязательно ознакомьтесь с общей информацией о врождённых мышечных дистрофиях / миопатиях. На этой странице вы найдёте различные публикации о диагностике ВМД, семейные и медицинские руководства.
Альтернативные названия мерозин-дефицитной врождённой мышечной дистрофии
- Мерозин-дефицитная врождённая мышечная дистрофия
- Мерозин-негативная врождённая мышечная дистрофия
- Врождённая мышечная дистрофия 1А типа (ВМД1А)
- LAMA2 Congenital Muscular Dystrophy (LAMA2-CMD)
- Merosin-deficient CMD (MDC1A)
На данной странице будет использоваться сокращение ВМД1А, в т.ч. в переводе иностранных материалов.
Ниже перевод материалов CureCMD.
Врождённая мышечная дистрофия (ВМД) 1А возникает из-за генетических мутаций белка мерозина – подвид белка ламинина, который находится в мышечных клетках. Пациенты с ВМД1А испытывают мышечную слабость, при ВМД1А развиваются контрактуры. Люди с ВМД1А могут родиться с полной или частичной недостаточностью мерозина в зависимости от генетических мутаций.
Характеристики врожденной мышечной дистрофии 1А
- Мышечная слабость
- Гипотония
- Мышечные контрактуры
- Ригидный позвоночник
- Сколиоз
- Дыхательная недостаточность, проблемы с кормлением
- Судороги
- Нарушения сердечной деятельности
- Патологии головного мозга
Физиология врожденной мышечной дистрофии 1А
ВМД1А возникает из-за двух мутаций в гене LAMA2 (laminin alpha 2), который продуцирует мерозин – один из ламининовых белков.
Мерозин составляет часть внеклеточного матрикса мембраны мышечных клеток. Внеклеточный матрикс образует внешнюю среду вокруг мышечной клетки.
Он выполняет критические функции, поддерживая стабильность мышечной клетки и её регенерацию, позволяя мышечной клетке прикрепляться к матриксу.
Ламинины играют важную роль в процессе связывания себя и других белков не только с внеклеточным матриксом, но и с мембраной мышечных клеток. Ламинины помогают поддерживать стабильность мышечных волокон.
Пациенты с ВМД1А рождаются с полным или частичным дефицитом мерозина в зависимости от того, где находятся генетические мутации.
У людей с полным отсутствием (дефицитом) мерозина заболевание начинает развиваться с самого рождения. Уже на ранней стадии жизни появляется мышечная гипотония (снижение мышечного тонуса или гибкости), прогрессируют контрактуры, возникают проблемы с дыханием и кормлением. Могут развиться сколиоз и ригидный позвоночник. У некоторых людей наблюдаются сердечные патологии.
У людей с частичным дефицитом мерозина заболевание может иметь более мягкое начало, либо проявиться позднее. При частичном дефиците мерозина заболевание протекает не так агрессивно, как при его полном отсутствии. Некоторые пациенты сохраняют способность ходить до взрослого возраста.
При ВМД1А могут наблюдаться аномалии белого вещества головного мозга, из-за чего у некоторых пациентов развиваются судороги. Такие аномалии выявляются при проведении МРТ-обследования головного мозга.
Наследование и генетическое тестирование
ВМД1А наследуется аутосомно-рецессивным способом, то есть должны присутствовать две патогенные мутации, вызывающие заболевание. Эти мутации могут быть унаследованы от каждого родителя или могут быть спонтанными (деново).
Поскольку ген LAMA2 является довольно большим, и исследователи все еще работают над его полной картой, довольно часто у людей с ВМД1А выявляют только одну из двух мутаций.
Тогда для подтверждения диагноза ВМД1А принимаются во внимание физические проявления заболевания, повышенный уровень креатинкиназы, изменения белого вещества головного мозга и частичное или полное отсутствие мерозина в мышечном биоптате мышц или кожи.
Диагностирование ВМД1А
Для диагностирования ВМД1А, помимо внешних признаков, характерных для этого заболевания, необходимо проводить анализ крови на уровень креатинин-киназы (КК).
Уровень КК у людей с ВМД1А часто значительно повышен, но поскольку повышенный уровень КК свойственен и другим подтипам ВМД, данный показатель можно использовать только для исключения других нервно-мышечных заболеваний. Можно также провести биопсию мышц или кожи, чтобы выявить, есть ли частичное или полное отсутствие ламинина.
Генетическое тестирование может дать окончательный диагноз, и этот анализ менее инвазивен по сравнению с биопсией. Секвенирование нового поколения в настоящее время широко доступно, поэтому многие врачи теперь предпочитают этот метод тестирования.
Диагностика ВМД1А
Общий список клиник и врачей, занимающихся нервно-мышечными заболеваниями.
На странице о врождённых мышечных дистрофиях вы можете ознакомиться с публикациями о диагностике ВМД.
Статьи по диагностике нервно-мышечных заболеваний
– МРТ мышц
– генетическая диагностика
Что важно знать при ВМД1А?
Ознакомьтесь с семейными и медицинскими руководствами по врождённым мышечным дистрофиям / миопатиям.
Если у вас есть руководства конкретно о ВМД1А, просим вас поделиться. Информацию присылайте нам на почту imio.letters@gmail.com.
Важно! Регистры (реестры) пациентов с ВМД1А
Международный регистр по врождённым мышечным заболеваниям (CMDIR – Congenital muscle disease international register).
Список регистров пациентов с ВМД1А в разных странах на сайте Treat-NMD.
Почему нужно регистрироваться в реестре?
По мере того, как разрабатываются новые препараты, появляется необходимость их тестирования в клинических условиях, и иногда требуются годы, чтобы найти необходимое количество пациентов для исследований, поскольку генетические нервно-мышечные заболевания являются редкими (орфанными) заболеваниями.
Для этого в разных странах ведутся реестры пациентов — базы данных по генетической и клинической информации о людях, страдающих от генетических нервно-мышечных заболеваний и желающих ускорить процесс исследований.
Реестр позволяет специалистам получить информацию о состоянии и количестве больных определённым заболеванием. Данная информация способствует развитию и улучшению стандартов лечения пациентов.
Он используется, чтобы найти участников для проведения клинических испытаний, а также помочь специалистам получить больше информации о заболевании.
Организации и сообщества, посвящённые ВМД1А
Статьи о ВМД на нашем сайте
Статьи о ВМД типа 1А на нашем сайте
")
")



